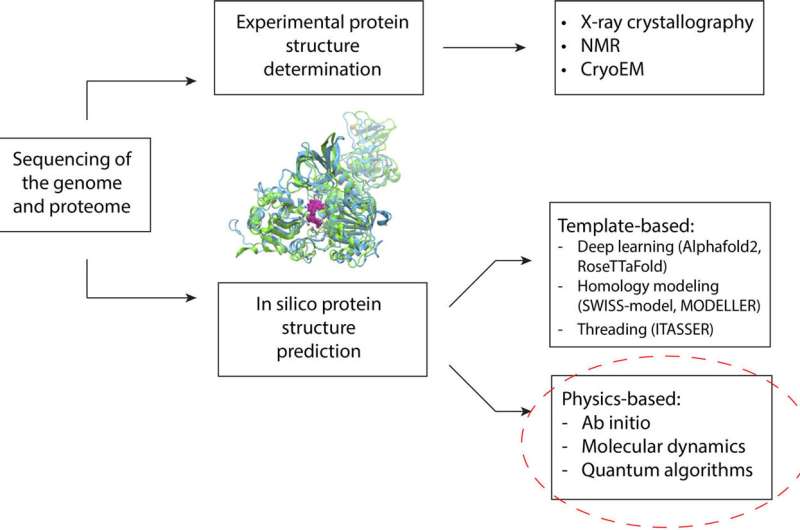
Des chercheurs de la Cleveland Clinic et d'IBM ont récemment publié leurs résultats dans le Journal of Chemical Theory and Computation. cela pourrait jeter les bases de l'application des méthodes informatiques quantiques à la prédiction de la structure des protéines.
Pendant des décennies, les chercheurs ont exploité des approches informatiques pour prédire les structures des protéines. Une protéine se replie dans une structure qui détermine son fonctionnement et se lie à d’autres molécules du corps. Ces structures déterminent de nombreux aspects de la santé humaine et des maladies.
En prédisant avec précision la structure d’une protéine, les chercheurs peuvent mieux comprendre comment les maladies se propagent et ainsi développer des thérapies efficaces. Bryan Raubenolt, boursier postdoctoral de la Cleveland Clinic, Ph.D. et le chercheur IBM Hakan Doga, Ph.D. a dirigé une équipe pour découvrir comment l'informatique quantique peut améliorer les méthodes actuelles.
Ces dernières années, les techniques d’apprentissage automatique ont fait des progrès significatifs dans la prédiction de la structure des protéines. Ces méthodes s'appuient sur des données d'entraînement (une base de données de structures protéiques déterminées expérimentalement) pour faire des prédictions. Cela signifie qu’ils sont limités par le nombre de protéines qu’on leur a appris à reconnaître. Cela peut conduire à des niveaux de précision inférieurs lorsque les programmes/algorithmes rencontrent une protéine mutée ou très différente de celles sur lesquelles ils ont été formés, ce qui est courant dans les troubles génétiques.
La méthode alternative consiste à simuler la physique du repliement des protéines. Les simulations permettent aux chercheurs d'examiner les différentes formes possibles d'une protéine donnée et de trouver la plus stable. La forme la plus stable est essentielle à la conception de médicaments.
Le défi est que ces simulations sont quasiment impossibles sur un ordinateur classique, au-delà d’une certaine taille de protéine. D'une certaine manière, augmenter la taille de la protéine cible est comparable à l'augmentation des dimensions d'un Rubik's cube. Pour une petite protéine contenant 100 acides aminés, un ordinateur classique aurait besoin d'un temps égal à l'âge de l'univers pour rechercher de manière exhaustive tous les résultats possibles, explique le Dr Raubenolt.
Pour aider à surmonter ces limites, l’équipe de recherche a appliqué un mélange de méthodes informatiques quantiques et classiques. Ce cadre pourrait permettre aux algorithmes quantiques d'aborder les domaines qui représentent un défi pour l'informatique classique de pointe, notamment la taille des protéines, le désordre intrinsèque, les mutations et la physique impliquée dans le repliement des protéines. Le cadre a été validé en prédisant avec précision le repliement d'un petit fragment d'une protéine du virus Zika sur un ordinateur quantique, par rapport aux méthodes classiques de pointe.
Les premiers résultats du cadre hybride quantique-classique ont surpassé à la fois une méthode basée sur la physique classique et AlphaFold2. Bien que ce dernier soit conçu pour fonctionner mieux avec des protéines plus grosses, il démontre néanmoins la capacité de ce cadre à créer des modèles précis sans s'appuyer directement sur des données d'entraînement substantielles.
Les chercheurs ont utilisé un algorithme quantique pour modéliser d'abord la conformation d'énergie la plus basse pour le squelette du fragment, ce qui constitue généralement l'étape de calcul la plus exigeante en termes de calcul. Des approches classiques ont ensuite été utilisées pour convertir les résultats obtenus par l'ordinateur quantique, reconstruire la protéine avec ses chaînes latérales et effectuer le raffinement final de la structure avec les champs de force de la mécanique moléculaire classique.
Le projet montre l'une des façons dont les problèmes peuvent être déconstruits en parties, avec des méthodes d'informatique quantique abordant certaines parties et l'informatique classique d'autres, pour une précision accrue.
"L'un des aspects les plus uniques de ce projet est le nombre de disciplines impliquées", explique le Dr Raubenolt. « L'expertise de notre équipe s'étend de la biologie et de la chimie computationnelles, de la biologie structurale, de l'ingénierie logicielle et de l'automatisation, à la physique atomique et nucléaire expérimentale, aux mathématiques et, bien sûr, à l'informatique quantique et à la conception d'algorithmes. Il a fallu les connaissances de chacun de ces domaines pour créer un cadre informatique capable d'imiter l'un des processus les plus importants de la vie humaine."
La combinaison de méthodes informatiques classiques et quantiques par l'équipe constitue une étape essentielle pour faire progresser notre compréhension des structures protéiques et de leur impact sur notre capacité à traiter et à prévenir les maladies. L'équipe prévoit de continuer à développer et à optimiser des algorithmes quantiques capables de prédire la structure de protéines plus grandes et plus sophistiquées.
"Ce travail constitue une étape importante dans l'exploration des domaines dans lesquels les capacités de calcul quantique pourraient démontrer leurs atouts en matière de prédiction de la structure des protéines", déclare le Dr Doga. "Notre objectif est de concevoir des algorithmes quantiques capables de prédire les structures des protéines de la manière la plus réaliste possible."
Plus d'informations : Hakan Doga et al, Une perspective sur la prédiction de la structure des protéines à l'aide d'ordinateurs quantiques, Journal of Chemical Theory and Computation (2024). DOI :10.1021/acs.jctc.4c00067
Fourni par la Cleveland Clinic