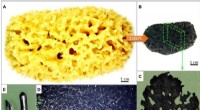
Les scientifiques ont développé un algorithme d'apprentissage automatique pour prédire la densité cristalline moléculaire 3D à partir de structures chimiques 2D. Crédit :Laboratoire national Lawrence Livermore
Un objectif de longue date par les chimistes de nombreuses industries, y compris l'énergie, médicaments, énergétique, additifs alimentaires et semi-conducteurs organiques, est d'imaginer la structure chimique d'une nouvelle molécule et de pouvoir prédire comment elle fonctionnera pour une application souhaitée. En pratique, cette vision est difficile, nécessitant souvent un travail de laboratoire approfondi pour synthétiser, isoler, purifier et caractériser des molécules nouvellement conçues pour obtenir les informations souhaitées.
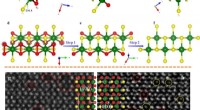
Récemment, une équipe de matériaux et d'informaticiens du Lawrence Livermore National Laboratory (LLNL) a concrétisé cette vision pour les molécules énergétiques en créant des modèles d'apprentissage automatique (ML) qui peuvent prédire les propriétés cristallines des molécules à partir de leurs seules structures chimiques, comme la densité moléculaire. Prédire les descripteurs de la structure cristalline (plutôt que la structure cristalline entière) offre une méthode efficace pour déduire les propriétés d'un matériau, accélérant ainsi la conception et la découverte de matériaux. La recherche apparaît dans le Journal of Chemical Information and Modelling .
"L'un des modèles ML les plus importants de l'équipe est capable de prédire la densité cristalline de molécules énergétiques et de type énergétique avec un degré élevé de précision par rapport aux méthodes précédentes basées sur la ML, " dit Phan Nguyen, Mathématicien appliqué du LLNL et co-premier auteur de l'article.
"Même par rapport à la théorie de la fonctionnelle de la densité (DFT), une méthode coûteuse en calcul et fondée sur la physique pour la prédiction de la structure cristalline et des propriétés cristallines, le modèle ML offre une précision compétitive tout en nécessitant une fraction du temps de calcul, " a déclaré Donald Loveland, Informaticien LLNL et co-premier auteur.
Les membres de la High Explosive Application Facility (HEAF) de LLNL ont déjà commencé à tirer parti de l'interface Web du modèle, dans le but de découvrir de nouveaux matériaux énergétiques insensibles. En entrant simplement la structure chimique 2D des molécules, Les chimistes de HEAF ont pu déterminer rapidement la densité cristalline prévue de ces molécules, ce qui est étroitement corrélé aux métriques de performance énergétique potentielles.
« Nous sommes ravis de voir les résultats de nos travaux être appliqués à des missions importantes du laboratoire. Ce travail contribuera certainement à accélérer la découverte et l'optimisation de nouveaux matériaux à l'avenir, " dit Yong Han, Scientifique des matériaux du LLNL et chercheur principal du projet.
Les efforts de suivi au sein de la Division des sciences des matériaux ont utilisé le modèle ML en conjonction avec un modèle génératif pour rechercher rapidement et efficacement de grands espaces chimiques pour des candidats à haute densité.
"Les deux efforts repoussent les limites de la découverte des matériaux et sont facilités par le nouveau paradigme de la fusion de la science des matériaux et de l'apprentissage automatique, " a déclaré Anna Hiszpanski, Scientifique des matériaux LLNL et auteur co-correspondant de l'article.
L'équipe continue de rechercher de nouvelles propriétés d'intérêt pour le laboratoire dans le but de fournir une suite de modèles prédictifs que les scientifiques des matériaux pourront utiliser dans leurs recherches.