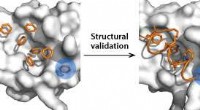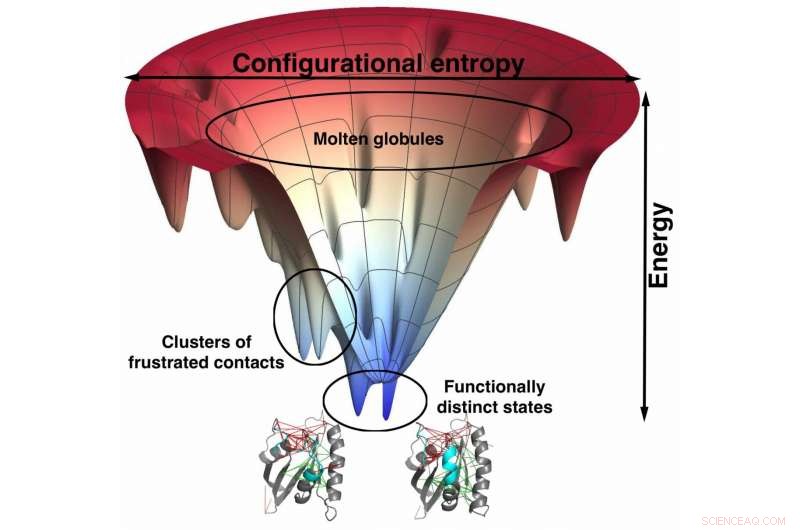
Les modèles à l'échelle atomique des scientifiques de l'Université Rice basés sur ceux utilisés pour prédire le repliement des protéines montrent une forte corrélation entre les sites de liaison peu frustrés et la spécificité du médicament. L'entonnoir, une représentation visuelle du paysage énergétique de la protéine lors de son repliement, aide à localiser ces sites frustrés. De tels modèles pourraient conduire à des médicaments mieux conçus avec moins d'effets secondaires. Crédit :Mingchen Chen/Université du riz
Savoir précisément où les protéines sont frustrées pourrait grandement contribuer à la fabrication de meilleurs médicaments.
C'est l'un des résultats d'une nouvelle étude menée par des scientifiques de l'Université Rice à la recherche des mécanismes qui stabilisent ou déstabilisent les sections clés des biomolécules.
Modèles à l'échelle atomique du théoricien de Rice Peter Wolynes, l'auteur principal et ancien élève Mingchen Chen et leurs collègues du Center for Theoretical Biological Physics montrent que non seulement certaines séquences frustrées spécifiques dans les protéines sont nécessaires pour leur permettre de fonctionner, leur localisation offre également des indices pour obtenir une meilleure spécificité des médicaments.
Cette connaissance pourrait également aider à concevoir des médicaments avec moins d'effets secondaires, dit Wolyne.
L'étude en libre accès de l'équipe apparaît dans Communication Nature .
Les modèles à l'échelle de l'atome se concentrent sur les interactions au sein des sites de liaison possibles plutôt que sur la grande majorité des interactions dans les protéines qui guident leur repliement. Les modèles de résolution plus fine permettent l'incorporation de co-facteurs comme des ligands chimiquement actifs, y compris les molécules médicamenteuses. Les chercheurs affirment que cette capacité donne un nouvel aperçu des raisons pour lesquelles les ligands ne sont mieux capturés que par des protéines spécifiques et non par d'autres.
"Ligands non naturels, " alias la drogue, ont tendance à mieux se lier avec ces poches frustrées dans les protéines qui deviennent peu frustrées une fois que les médicaments se lient, dit Wolyne. Avoir un moyen de trouver et d'apprendre les détails de ces sites peu frustrés aiderait les sociétés pharmaceutiques à éliminer beaucoup d'essais et d'erreurs.
"La façon standard de concevoir un médicament est d'en essayer 10, 000 sites de liaison sur une protéine pour trouver ceux qui conviennent, " dit Wolynes. " Nous disons que vous n'avez pas à échantillonner tous les sites de liaison possibles, juste un nombre raisonnablement juste pour comprendre les statistiques de ce qui pourrait fonctionner dans les environnements locaux.
"C'est la différence entre participer à un sondage et organiser une élection, " dit-il. " Le sondage est moins cher, mais vous devrez quand même vérifier les choses."
Les chercheurs de Rice sont connus pour leur théorie du paysage énergétique sur la façon dont les protéines se replient. Il utilise généralement des modèles à gros grains dans lesquels les acides aminés ne sont représentés que par quelques sites.
Cette stratégie prend moins de puissance de calcul que d'essayer de déterminer les positions dans le temps de chaque atome dans chaque résidu, et pourtant, il s'est avéré très précis pour prédire comment les protéines se replient en fonction de leurs séquences. Mais pour cette étude, les chercheurs ont modélisé des protéines et des complexes protéine-ligand au niveau atomique pour voir s'ils pouvaient trouver comment la frustration donne à certaines parties d'une protéine la flexibilité nécessaire pour se lier à d'autres molécules.
"L'un des avantages de la modélisation à la résolution de tous les atomes est qu'elle nous permet d'évaluer si les molécules médicamenteuses s'intègrent bien dans les sites de liaison ou non, " Wolynes a déclaré. "Cette méthode est capable de montrer rapidement si un site de liaison pour un certain médicament sera peu frustré ou restera une région frustrée. Si après la liaison de la molécule, le site reste frustré, la protéine pourrait se réorganiser ou le médicament pourrait changer son orientation de telle manière qu'il pourrait provoquer des effets secondaires. »
La modélisation des sites frustrés - et parfois leur modification pour voir ce qui se passerait - permet aux chercheurs de voir comment la spécificité du médicament est en corrélation avec les poches de liaison. Analyse de frustration, ils ont écrit, fournit "une voie pour le criblage de composés plus spécifiques pour la découverte de médicaments".
« Cette notion de frustration était présente au tout début de nos travaux sur le repliement des protéines, " dit Wolynes. " Quand nous l'avons appliqué à de vraies molécules de protéines, nous avons trouvé des exemples où le mécanisme de pliage a violé ce que nous pourrions prédire à partir d'un entonnoir parfait. Ensuite, nous avons découvert que ces écarts par rapport à l'image de l'entonnoir se produisaient là où se trouvait la protéine, En réalité, quelque peu frustré.
"C'était comme l'exception qui confirme la règle, " dit-il. " Quelque chose qui est vrai tout le temps peut être insignifiant. Mais si ce n'est pas vrai 1% du temps, c'est un problème à résoudre, et nous avons pu le faire avec AWSEM, notre logiciel de prédiction de structure."
Etendre le logiciel pour analyser la frustration au niveau atomique est possible, comme décrit par le groupe dans un autre article récent. Mais le coût de calcul du suivi de chaque atome d'une protéine est si élevé que les chercheurs avaient besoin d'un moyen d'échantillonner les mouvements de régions spécifiques où la frustration pourrait perturber la voie de repliement.
"Mingchen s'est rendu compte qu'il existait un algorithme efficace pour échantillonner les environnements locaux dans les sites de liaison tout en conservant la résolution atomistique, " dit Wolynes, qui a noté lui et Chen, maintenant dans l'industrie privée, utilisent les modèles pour étudier les thérapies possibles, y compris les médicaments liés au COVID-19.