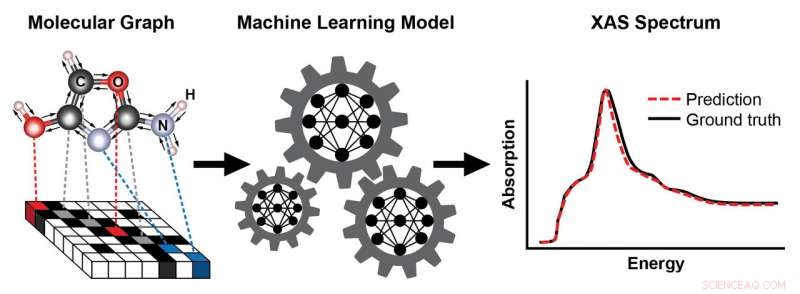
Schéma montrant les étapes d'entraînement d'un modèle d'apprentissage automatique pour prédire un spectre d'absorption des rayons X (XAS) basé sur la structure connue d'une molécule. La structure de la molécule est représentée sous forme de graphique, avec des atomes comme nœuds et des liaisons chimiques comme arêtes. Cette représentation capture la connectivité des atomes - ici, carbone (C), l'oxygène (O), azote (N), et l'hydrogène (H)—et le type et la longueur des liaisons chimiques qui les relient. Le spectre XAS résultant contient des informations riches sur l'environnement chimique local des atomes absorbants, tels que leur symétrie et le nombre d'atomes voisins. Crédit :Laboratoire national de Brookhaven
La spectroscopie d'absorption des rayons X (XAS) est une technique de caractérisation populaire pour sonder la structure atomique locale et les propriétés électroniques des matériaux et des molécules. Parce que les atomes de chaque élément absorbent les rayons X à des énergies caractéristiques, XAS est bien adapté pour cartographier la distribution spatiale des éléments dans un échantillon. Typiquement, les scientifiques effectuent des expériences XAS sur des sources de lumière synchrotron, telles que la National Synchrotron Light Source II (NSLS-II), car elles fournissent des rayons X réglables. En mesurant l'absorbance dans un échantillon à différentes énergies de rayons X, les scientifiques peuvent générer un graphique appelé spectre d'absorption des rayons X.
« XAS est une capacité clé pour les utilisateurs du NSLS-II du Brookhaven National Laboratory et du Center for Functional Nanomaterials (CFN), les deux installations des utilisateurs du bureau des sciences du département de l'Énergie des États-Unis (DOE) qui sont ouvertes à la communauté de la recherche scientifique, " dit Deyu Lu, un physicien dans le groupe de théorie et de calcul du CFN. "Avec les bons outils d'analyse, XAS peut fournir des informations considérables sur la recherche en nanosciences. Le développement de tels outils est au cœur de notre mission en tant qu'installations utilisateurs."
Classification des environnements chimiques locaux
Différentes régions du spectre d'absorption des rayons X sont sensibles à différents aspects des propriétés du matériau dans un échantillon. Par exemple, la structure de bord proche d'absorption de rayons X (XANES) se concentre sur la région de bord proche du spectre, juste au-dessus de l'énergie initiale suffisante pour exciter un électron des couches internes d'un atome vers un état vide. XANES code des informations riches sur l'environnement chimique local des atomes absorbants dans un échantillon, y compris leur coordination géométrique, symétrie, et l'état de charge (le nombre d'électrons gagnés ou perdus par la liaison chimique). Mais l'analyse des données spectrales est très difficile en raison de leur nature abstraite.
"Contrairement à une image au microscope d'un matériau où vous pouvez voir directement des caractéristiques telles que la cristallinité ou les défauts, Les spectres XANES encodent des informations qui nécessitent une expertise du domaine pour interpréter, " expliqua Lu.
L'interprétation standard des signaux dans un spectre XANES repose sur des caractéristiques connues sous le nom d'« empreintes digitales, " qui sont construits à partir de mesures sur des matériaux de référence. Cependant, cette approche par empreinte digitale échoue lorsque l'échantillon n'est pas un simple cristal et que les matériaux de référence pertinents ne peuvent pas être facilement identifiés.
Des simulations théoriques à grande échelle à partir de modèles de structure atomique peuvent fournir des informations très utiles pour l'interprétation des spectres XANES expérimentaux ; cependant, ces simulations sont souvent coûteuses en temps de calcul et chronophages, et leur niveau de précision dépend fortement des approximations théoriques choisies et du système à l'étude. Par conséquent, une interprétation spectrale robuste est actuellement le goulot d'étranglement des études XAS. Par ailleurs, L'interprétation en temps réel des spectres XAS est apparue comme un nouveau défi pour les études de l'évolution dynamique des matériaux en conditions opératoires et l'expérimentation autonome. Le besoin de robustesse, l'interprétation spectrale efficace est de plus en plus répandue dans les sources lumineuses synchrotron.
"Temps réel, interprétation précise de la diffusion des rayons X et des mesures de spectroscopie telles que l'absorption des rayons X, fluorescence, et la diffraction est une capacité importante pour les utilisateurs effectuant des recherches à NSLS-II et d'autres installations de lumière synchrotron, " a déclaré Mehmet Topsakal, un associé scientifique au sein du groupe Matériaux pour les applications énergétiques du Département des sciences et technologies nucléaires de Brookhaven, qui développe des techniques avancées d'analyse de données et d'apprentissage automatique pour la spectroscopie des rayons X. "Chaque année, des milliers de scientifiques du monde entier viennent à NSLS-II pour sonder les propriétés de divers matériaux. Un pipeline d'analyse spectrale de pointe permettrait aux utilisateurs d'obtenir des commentaires utiles sur leurs échantillons pendant que les expériences sont en cours et d'effectuer des ajustements à la volée pour guider les expériences. La question est, comment pouvons-nous faire une interprétation spectrale en temps réel pour découvrir des corrélations structure-spectre ?"
Extraire des informations avec l'apprentissage automatique
Tirer parti du big data et du machine learning, Lu et Topsakal ont décidé de répondre à cette question avec le scientifique informaticien Shinjae Yoo de la Computational Science Initiative (CSI) de Brookhaven Lab et le doctorat de l'Université Columbia. candidat et chercheur diplômé en sciences informatiques du DOE Matthew Carbone.
"La bourse d'études supérieures en sciences informatiques du DOE m'a offert une opportunité unique d'aller au-delà de mes recherches de doctorat en physique chimique à Columbia pour explorer la puissance des algorithmes d'apprentissage automatique, travailler aux côtés des scientifiques de Brookhaven, " a déclaré Carbone. " L'apprentissage automatique s'appuie sur des ensembles de données massifs pour créer des modèles très perspicaces qui, une fois formé, peut faire des prédictions à la volée sur de nouvelles données. De tels modèles pourraient être utilisés pour contourner les calculs coûteux de chimie quantique et prendre en charge la caractérisation des matériaux operando. »
Les membres de cette équipe et les collaborateurs travaillent depuis plusieurs années sur des mappages spectre-structure et structure-spectre. En 2017, ils ont développé des modèles d'apprentissage automatique pour prédire les nombres de coordination moyens de nanoparticules métalliques à partir des spectres XANES. L'année dernière, ils ont créé une base de données XANES pour résoudre la structure locale d'un revêtement d'oxyde de titane amorphe pour des applications photocatalytiques. Ils ont également construit un modèle d'apprentissage automatique capable de prédire la symétrie locale des atomes absorbants à partir de spectres XANES simulés d'oxydes de métaux de transition.
"Lors de l'interprétation spectrale basée sur l'expertise du domaine, nous avons tendance à nous concentrer sur des fonctionnalités spécifiques conçues à partir de notre intuition, " a déclaré Lu. " L'apprentissage automatique peut extraire les informations dont nous avons besoin d'une manière statistiquement importante qui élimine les biais humains. "
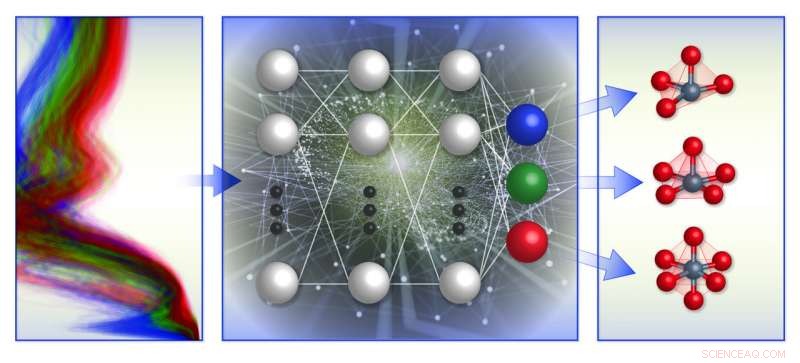
Une illustration schématique du cadre de classification de l'environnement chimique local basé sur le spectre de l'équipe. Ils ont formé des modèles d'apprentissage automatique (au milieu) avec une base de données de spectres d'absorption des rayons X (à gauche) pour prédire la géométrie locale autour des ions de métaux de transition chargés positivement (à droite). Crédit :Laboratoire national de Brookhaven
Prédiction des spectres d'absorption des rayons X
Forts de leurs succès passés, l'équipe s'est attaquée à un problème plus difficile :entraîner un modèle d'apprentissage automatique pour prédire rapidement des spectres basés sur des structures moléculaires connues. Un tel modèle éviterait le besoin de simulations coûteuses en calculs, qui ne sont pas réalisables lors des expériences operando, lorsque les scientifiques étudient des matériaux dans des conditions d'exploitation. Malgré les efforts croissants d'apprentissage automatique pour prédire les propriétés chimiques des matériaux, les prédictions directes des fonctions spectrales de matériaux réels n'avaient pas encore été réalisées.
« Une difficulté technique consiste à créer une représentation optimale des structures moléculaires pouvant coder la symétrie inhérente des molécules en tant que caractéristiques d'entrée pour le modèle d'apprentissage automatique, " dit Yoo.
Adoptant une idée récente proposée par des scientifiques de Google, Topsakal et Carbone ont construit un modèle d'apprentissage automatique basé sur une représentation graphique des molécules en entrée, où les atomes sont représentés comme des nœuds et les liaisons chimiques comme des arêtes.
"Les ordinateurs ne peuvent pas voir les molécules comme nous, " a déclaré Topsakal. " Un graphique est un moyen naturel de coder la structure et la connectivité d'une molécule, capturant les atomes qui sont connectés et le type et la longueur des liaisons chimiques qui les relient. De plus, cette représentation est invariante aux transformations telles que les translations et les rotations. Ce concept est analogue à celui de la reconnaissance d'images, où un objet tel qu'un chat ou un chien en arrière-plan peut toujours être classé correctement une fois l'image transformée."
Pour entraîner le modèle pour une démonstration de preuve de principe, l'équipe a utilisé une base de données bien établie (appelée QM9) contenant des informations structurelles et chimiques calculées sur 134, 000 petites molécules avec jusqu'à neuf atomes lourds par type d'atome (carbone, azote, oxygène, et fluor). A partir de cette base de données, ils ont sélectionné deux sous-ensembles d'entraînement - un sous-ensemble avec des molécules contenant au moins un atome d'oxygène, et un autre sous-ensemble avec des molécules contenant au moins un atome d'azote et calculé leurs spectres XANES correspondants. Puis, ils ont utilisé leurs modèles entraînés pour prédire les spectres XANES pour les bords d'absorption d'oxygène et d'azote correspondant aux excitations d'électrons dans la couche la plus interne des atomes respectifs.
Le modèle d'apprentissage automatique a reproduit presque tous les pics d'absorption significatifs et a prédit les positions des pics (énergies auxquelles les pics apparaissent) et les hauteurs (intensités d'absorption) avec une très grande précision. Le modèle a également automatiquement pris en compte la connaissance du domaine selon laquelle la spectroscopie d'absorption des rayons X est sensible aux groupes fonctionnels, ou des groupes d'atomes ayant des propriétés chimiques et une réactivité similaires. Selon le groupe fonctionnel auquel appartient l'atome absorbant, différentes caractéristiques apparaissent dans les spectres.
« Nous sommes les premiers à démontrer qu'un modèle d'apprentissage automatique peut être utilisé pour prédire avec précision les fonctions spectrales complètes de systèmes physiques réels directement à partir de leurs structures, " a déclaré Topsakal. " Bien que nous nous soyons concentrés sur la spectroscopie d'absorption des rayons X dans notre étude, cette méthode pourrait être généralisée pour prédire les informations spectrales pour d'autres techniques populaires, y compris la spectroscopie infrarouge et gamma.
"Une fois que nous avons formé le modèle d'apprentissage automatique, nous n'avons pas besoin de faire des simulations physiques chronophages, qui prend quelques minutes, les heures, voire des jours, " a déclaré Yoo. " Nous avons permis non seulement la prédiction de spectres en temps réel, mais également la génération simultanée de centaines et de milliers d'inférences de spectres en utilisant plusieurs unités de traitement graphique, ou GPU. Une telle technologie est essentielle pour permettre des contrôles automatisés des lignes de lumière et accélérer les découvertes scientifiques. Combiné avec des méthodes pour échantillonner des structures matérielles, de tels modèles peuvent être utilisés pour filtrer rapidement les structures pertinentes afin de favoriser la conception et la découverte de matériaux. »
Prochain, l'équipe souhaite combiner les concepts de leur modèle qui prédit la symétrie locale à partir des spectres XANES et ce nouveau modèle qui prédit les spectres XANES à partir des structures moléculaires. Finalement, leur objectif est d'extraire des informations plus complètes sur l'environnement chimique local ou même la structure de molécules entières à partir de mesures expérimentales.
"Outils d'apprentissage automatique, tels que ceux pour la reconnaissance d'images et de la parole et la découverte de médicaments, sont en plein développement, " a déclaré Lu. " La clé est de trouver comment adapter ces outils de manière innovante pour résoudre les problèmes de la science des matériaux. "
« Notre objectif dans le développement de technologies d'intelligence artificielle et d'apprentissage automatique est de résoudre des défis scientifiques uniques en adoptant les dernières avancées technologiques dans ces domaines et en proposant de nouvelles approches qui contribuent aux communautés de recherche respectives, " a ajouté Yoo.