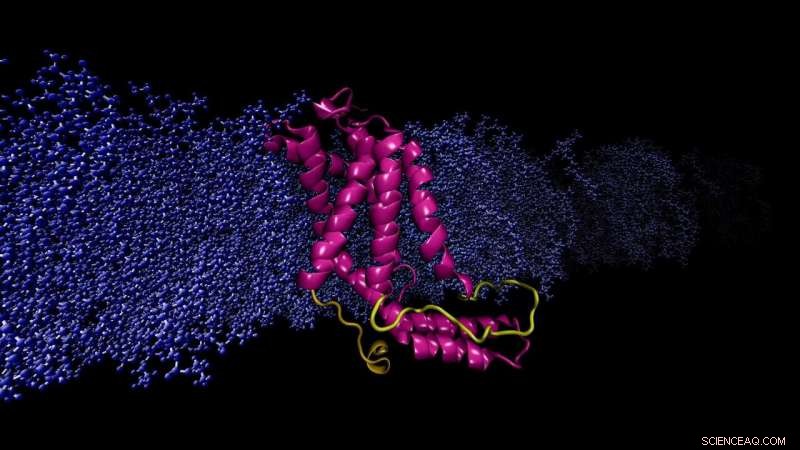
Membrane cellulaire incorporée dans la protéine YidC2 du modèle informatique. La boucle modélisée (jaune), manquant dans la structure cristalline aux rayons X, est crucial pour la stabilisation des protéines. Crédit :Sogol Moradi
Une nouvelle étude menée par des chimistes de l'Université de l'Arkansas montre que la cristallographie aux rayons X, la méthode standard de détermination de la structure des protéines, peut fournir des informations inexactes sur un ensemble critique de protéines, celles trouvées dans les membranes cellulaires, ce qui pourrait à son tour conduire à une conception de médicaments médiocre et inefficace.
Les résultats des chercheurs ont été publiés aujourd'hui dans Rapports scientifiques , une publication Nature.
"Les deux tiers de toutes les drogues, y compris ceux utilisés pour la chimiothérapie, protéines cibles présentes sur les membranes cellulaires, " dit Mahmoud Moradi, professeur adjoint de chimie et de biochimie au J. William Fulbright College of Arts and Sciences. "Malheureusement, Cristallographie aux rayons X, l'étalon-or pour déterminer la structure des protéines, a de nombreuses limitations lorsqu'il s'agit de ceux trouvés dans la membrane cellulaire. Notre travail expose, et à bien des égards, explique ces limites.
Considéré comme les molécules de cheval de bataille des cellules, les protéines sont responsables de presque toutes les tâches dans les systèmes vivants. Certaines protéines vivent à l'intérieur des cellules, et certains résident sur la membrane de la cellule, une couche externe de lipides qui sépare la cellule de son environnement extérieur. Les protéines membranaires sont d'une importance critique car elles régulent l'échange d'informations et de matériaux entre la cellule et son environnement, une tâche vitale pour la survie et le fonctionnement normal de la cellule, car tout trouble de la fonction protéique peut entraîner une maladie.
L'étude de la fonction des protéines est nécessaire pour comprendre la base moléculaire de la maladie. Pour faire ça, les chercheurs se sont appuyés sur la cristallographie aux rayons X, l'outil principal pour déterminer la forme et la structure des protéines. La cristallographie aux rayons X est également essentielle dans le but de concevoir des médicaments qui manipulent efficacement la fonction des protéines. Cependant, l'étude de la structure des protéines membranaires est difficile car leur environnement natif n'est pas compatible avec la cristallographie aux rayons X. Les chercheurs doivent retirer les protéines de leur environnement natif et les placer dans un environnement lipidique artificiel avant d'appliquer la technique.
Moradi et Thomas Harkey, un étudiant de premier cycle à l'époque et maintenant étudiant en médecine à l'Université de l'Arkansas pour les sciences médicales, ont abordé ce problème sous un angle différent. Depuis environ deux ans, ils ont utilisé un superordinateur du centre de calcul haute performance de l'Arkansas pour fonctionner en continu, calculs de niveau microseconde simulant la dynamique moléculaire de YidC2, une protéine membranaire avec une boucle cytoplasmique non résolue cristallographiquement dans sa structure moléculaire. Les boucles cytoplasmiques sont connues pour avoir une signification fonctionnelle dans les protéines membranaires.
Les simulations de Moradi et Harkey ont démontré que la boucle cytoplasmique de YidC2 stabilisait la protéine entière, en particulier la région C1, un domaine potentiellement important pour la conception de médicaments. Des groupes de tête de lipides hautement polaires ou chargés interagissaient avec et stabilisaient la boucle. Cette découverte a démontré que les boucles non résolues des protéines membranaires pourraient être importantes pour la stabilisation des protéines, malgré le manque apparent de structure moléculaire.
"Typiquement, si une partie d'une protéine n'est pas résolue en cristallographie aux rayons X, il est interprété comme dépourvu d'une structure particulière, " Moradi a déclaré. "Nous montrons que pour les protéines membranaires et en particulier les parties de la protéine qui interagissent avec la membrane cellulaire, cette interprétation n'est pas exacte et pourrait être trompeuse. Nous pensons que l'explication alternative de la maladie pourrait être que la protéine n'est pas étudiée dans son environnement membranaire natif."
Moradi a déclaré que leurs résultats ont également démontré que la chimie computationnelle et la technologie de superinformatique peuvent être utilisées pour modéliser les protéines membranaires avec plus de précision dans un environnement qui imite leur environnement physiologique.