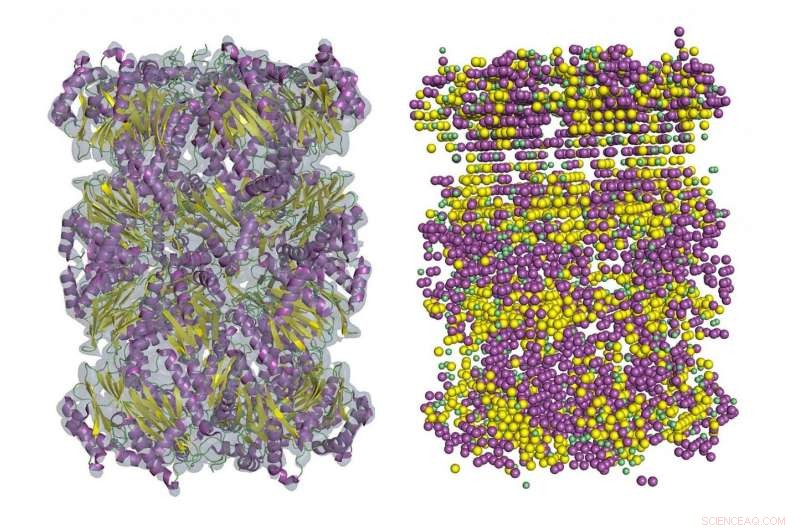
Un exemple de détection de structure secondaire dans une carte de densité cryo-EM à l'aide d'Emap2Sec. A gauche, une carte EM du protéasome 20S des archées (ID EMDB :EMD-1733). A droite sont détectées des structures secondaires par Emap2Sec. Les points en magenta sont les positions des hélices alpha détectées; les points jaunes sont des brins bêta détectés, et les points verts sont pour les bobines détectées (autres structures). Crédit :Image de l'Université Purdue/Daisuke Kihara
La microscopie cryoélectronique est maintenant la méthode la plus populaire pour déterminer les structures des protéines, qui aide les chercheurs à développer des médicaments pour différents types de maladies. Au cours des dernières décennies, il a remplacé la cristallographie aux rayons X car il peut imager des protéines qui ne peuvent pas facilement être transformées en gros cristaux. La nouvelle technique était si révolutionnaire qu'elle a valu à ses développeurs le prix Nobel de chimie 2017.
Le produit final de cryo-EM est une carte de la densité des atomes dans les molécules biologiques, mais pour atteindre le niveau de détail dont les chercheurs ont besoin, ils doivent mener une analyse plus approfondie. Une nouvelle étude dans la revue Méthodes naturelles décrit une technique pour amener les cartes à faible résolution à la hauteur.
L'approche utilisée par les chercheurs pour ce faire dépend du niveau de détail avec lequel ils commencent. Cartes à 2 à 3 ångström (Å, une unité de longueur utilisée pour exprimer la taille des atomes et des molécules) sont généralement considérés comme haute résolution. Cependant, des cartes de cette qualité sont difficiles à réaliser, et beaucoup sont encore couramment produits dans la gamme de 4 à 10 Å. De toutes les protéines déposées à la Banque de données de microscopie électronique de 2016 à 2018, plus de 50 % ont été résolus à une résolution intermédiaire.
"Si la résolution est meilleure que trois, alors les outils conventionnels peuvent tracer la position des acides aminés et construire une carte des positions des atomes. Mais fréquemment cryo-EM ne peut pas vous donner une carte de 3 Å, " dit Daisuke Kihara, professeur de sciences biologiques et d'informatique à l'Université Purdue. "Dans les cartes de 5 ou moins, vous ne pouvez généralement pas voir la connectivité de la chaîne du tout."
Les protéines sont en fait des chaînes d'acides aminés, et la liaison entre les groupes amino et les groupes carboxyle crée parfois certains modèles de repliement. Ces modèles, appelés hélices alpha et brins bêta, forment la structure secondaire de la protéine.
Dans les cartes de 5 à 8 Å, certains fragments de la structure secondaire des protéines sont généralement visibles, mais retracer toute la chaîne serait très difficile. La nouvelle méthode de Kihara, connu sous le nom d'Emap2sec, découvre des structures secondaires dans des cartes de 6 à 10 Å.
Emap2sec possède un réseau neuronal convolutif profond au cœur de son algorithme. Ces réseaux sont des systèmes d'apprentissage en profondeur principalement utilisés pour classer les images, les regrouper par similarité et effectuer une reconnaissance d'objet. Cela fonctionne pour l'identification de la structure des protéines dans les cartes 3D car la méthode "convolue" les caractéristiques de densité de la carte locale aux images d'une région plus grande lorsque les informations passent à travers les couches du réseau neuronal. La prédiction locale est faite dans le contexte d'une grande région de la carte.
Les structures secondaires identifiées dans les cartes 3D aident les chercheurs à attribuer des structures connues de protéines qui ont déjà été résolues dans la carte. Cela signifie qu'ils ont parfois un point de départ, ou au moins un indice de ce à quoi ressemble une partie de la structure. Emap2sec peut aider les chercheurs à insérer leur pièce dans le puzzle plus rapidement et plus facilement. Les informations de structure identifiées peuvent également être utiles pour trouver des erreurs dans la modélisation de la structure.