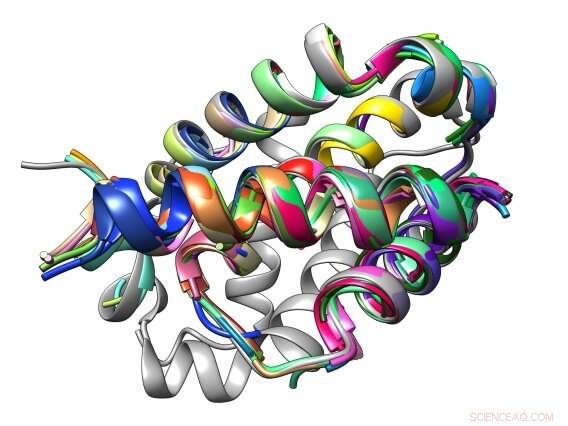
L'interface de liaison entre un peptide et sa protéine cible Bcl-2 est composée de motifs structuraux communs appelés TERM. Crédit :Sebastian Swanson et Avi Singer
Une façon de sonder des systèmes biologiques complexes est d'empêcher leurs composants d'interagir et de voir ce qui se passe. Cette méthode permet aux chercheurs de mieux comprendre les processus et fonctions cellulaires, augmenter les expériences quotidiennes en laboratoire, tests diagnostiques, et interventions thérapeutiques. Par conséquent, les réactifs qui empêchent les interactions entre les protéines sont très demandés. Mais avant que les scientifiques puissent rapidement générer leurs propres molécules personnalisées capables de le faire, ils doivent d'abord analyser la relation compliquée entre séquence et structure.
Les petites molécules peuvent entrer facilement dans les cellules, mais l'interface où deux protéines se lient l'une à l'autre est souvent trop grande ou manque des minuscules cavités requises pour que ces molécules ciblent. Les anticorps et les nanocorps se lient à de plus longues étendues de protéines, ce qui les rend mieux adaptés pour entraver les interactions protéine-protéine, mais leur grande taille et leur structure complexe les rendent difficiles à délivrer et instables dans le cytoplasme. Par contre, de courtes portions d'acides aminés, connu sous le nom de peptides, sont assez grands pour lier de longues étendues de protéines tout en étant assez petits pour entrer dans les cellules.
Le laboratoire Keating du département de biologie du MIT travaille dur pour développer des moyens de concevoir rapidement des peptides qui peuvent perturber les interactions protéine-protéine impliquant les protéines Bcl-2, qui favorisent la croissance du cancer. Leur approche la plus récente utilise un programme informatique appelé dTERMen, développé par un ancien du laboratoire Keating, Gevorg Grigoryan Ph.D. '07, actuellement professeur agrégé d'informatique et professeur agrégé adjoint de sciences biologiques et de chimie au Dartmouth College. Les chercheurs nourrissent simplement le programme de leurs structures souhaitées, et il crache des séquences d'acides aminés pour des peptides capables de perturber des interactions protéine-protéine spécifiques.
"C'est une approche tellement simple à utiliser, " dit Keating, un professeur de biologie du MIT et auteur principal de l'étude. "En théorie, vous pouvez mettre n'importe quelle structure et résoudre une séquence. Dans notre étude, le programme a proposé de nouvelles combinaisons de séquences qui ne ressemblent à rien de ce que l'on trouve dans la nature - il en a déduit une manière tout à fait unique de résoudre le problème. C'est excitant de découvrir de nouveaux territoires de l'univers de la séquence."
L'ancien postdoctorant Vincent Frappier et Justin Jenson Ph.D. '18 sont co-premiers auteurs de l'étude, qui paraît dans le dernier numéro de Structure .
Même problème, une approche différente
Jenson, pour sa part, a relevé le défi de concevoir des peptides qui se lient aux protéines Bcl-2 en utilisant trois approches distinctes. La méthode basée sur dTERMen, il dit, est de loin le plus efficace et le plus général qu'il ait essayé à ce jour.
Les approches standard pour découvrir les inhibiteurs peptidiques impliquent souvent la modélisation de molécules entières jusqu'à la physique et la chimie derrière les atomes individuels et leurs forces. D'autres méthodes nécessitent des criblages chronophages pour les meilleurs candidats de liaison. Dans les deux cas, le processus est ardu et le taux de réussite est faible.
dTERMens, par contre, ne nécessite ni physique ni criblage expérimental, et exploite des unités communes de structures protéiques connues, comme les hélices alpha et les brins bêta – appelés motifs structuraux tertiaires ou « TERM » – qui sont compilés dans des collections comme la Protein Data Bank. dTERMen extrait ces éléments structuraux de la banque de données et les utilise pour calculer quelles séquences d'acides aminés peuvent adopter une structure capable de se lier à et d'interrompre des interactions protéine-protéine spécifiques. Il faut une seule journée pour construire le modèle, et quelques secondes pour évaluer un millier de séquences ou concevoir un nouveau peptide.
"dTERMen nous permet de trouver des séquences susceptibles d'avoir les propriétés de liaison que nous recherchons, dans un robuste, efficace, et de manière générale avec un taux de réussite élevé, " dit Jenson. " Les approches passées ont pris des années. Mais en utilisant dTERMen, nous sommes passés des structures aux conceptions validées en quelques semaines."
Sur les 17 peptides qu'ils ont construits en utilisant les séquences conçues, 15 lié avec une affinité de type natif, perturbant les interactions protéine-protéine Bcl-2 qui sont notoirement difficiles à cibler. Dans certains cas, leurs conceptions étaient étonnamment sélectives et liées à un seul membre de la famille Bcl-2 par rapport aux autres. Les séquences conçues s'écartaient des séquences connues trouvées dans la nature, ce qui augmente considérablement le nombre de peptides possibles.
« Cette méthode permet une certaine flexibilité, " dit Frappier. " dTERMen est plus robuste au changement structurel, ce qui nous permet d'explorer de nouveaux types de structures et de diversifier notre portefeuille de candidats potentiels à la liaison."
Sonder l'univers des séquences
Compte tenu des avantages thérapeutiques de l'inhibition de la fonction Bcl-2 et du ralentissement de la croissance tumorale, le laboratoire de Keating a déjà commencé à étendre ses calculs de conception à d'autres membres de la famille Bcl-2. Ils ont l'intention de développer à terme de nouvelles protéines qui adoptent des structures jamais vues auparavant.
« Nous avons maintenant vu suffisamment d'exemples de diverses structures protéiques locales pour que les modèles informatiques des relations séquence-structure puissent être déduits directement des données structurelles, plutôt que de devoir être redécouvert à chaque fois des principes d'interaction atomistique, " dit Grigorian, Créateur de dTERMen. « C'est extrêmement excitant qu'une telle inférence basée sur la structure fonctionne et soit suffisamment précise pour permettre une conception robuste des protéines. Elle fournit un outil fondamentalement différent pour aider à résoudre les problèmes clés de la biologie structurelle, de la conception des protéines à la prédiction de la structure. »
Frappier espère un jour pouvoir cribler l'ensemble du protéome humain par ordinateur, en utilisant des méthodes telles que dTERMen pour générer des peptides de liaison candidats. Jenson suggère que l'utilisation de dTERMen en combinaison avec des approches plus traditionnelles de refonte des séquences pourrait amplifier un outil déjà puissant, permettre aux chercheurs de produire ces peptides ciblés. Idéalement, il dit, un jour, développer des peptides qui se lient et inhibent votre protéine préférée pourrait être aussi simple que d'exécuter un programme informatique, ou aussi courante que la conception d'une amorce d'ADN.
Selon Keating, bien que ce temps soit encore dans le futur, "notre étude est la première étape vers la démonstration de cette capacité sur un problème de portée modeste."
Cette histoire est republiée avec l'aimable autorisation de MIT News (web.mit.edu/newsoffice/), un site populaire qui couvre l'actualité de la recherche du MIT, innovation et enseignement.