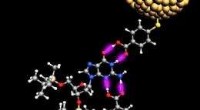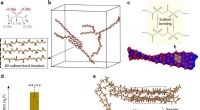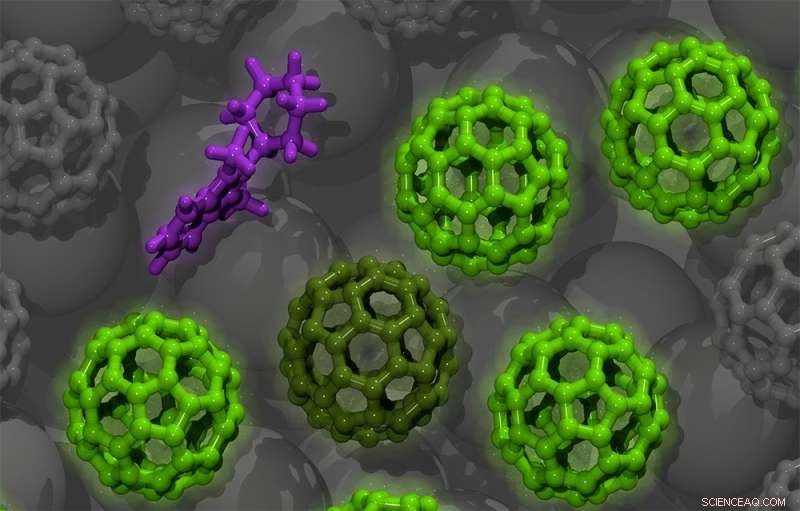
Illustration d'un semi-conducteur organique dopé à base de molécules de fullerène C60 (vert). Le dopant benzimidazoline (violet) donne un électron aux molécules C60 dans son environnement (vert foncé). Ces électrons peuvent alors se propager à travers le matériau semi-conducteur (vert clair). Crédit :S. Hutsch/F. Ortmann, TU Dresde
Semi-conducteurs, une classe de matériaux pouvant servir à la fois de conducteur électrique et d'isolant selon les circonstances, sont fondamentaux pour l'électronique moderne. Le silicium est le semi-conducteur le plus utilisé, mais ces dernières années, les chercheurs ont étudié une plus grande gamme de matériaux, y compris des molécules qui peuvent être adaptées pour répondre à des besoins électroniques spécifiques.
Les supercalculateurs sont des outils de recherche indispensables pour étudier les matériaux semi-conducteurs complexes à un niveau fondamental. Récemment, une équipe de scientifiques de la TU Dresden a utilisé le supercalculateur SuperMUC du Leibniz Supercomputing Center pour affiner sa méthode d'étude des semi-conducteurs organiques. L'équipe utilise une approche appelée dopage des semi-conducteurs, un processus dans lequel des impuretés sont intentionnellement introduites dans un matériau pour lui conférer des propriétés semi-conductrices spécifiques. Il a récemment publié ses résultats dans Matériaux naturels .
« Les semi-conducteurs organiques commencent à être utilisés dans de nouveaux concepts d'appareils, " a déclaré le chef d'équipe Dr Frank Ortmann. " Certains d'entre eux sont déjà sur le marché, mais certains sont encore limités par leur inefficacité. Nous recherchons des mécanismes de dopage, une technologie clé pour le réglage des propriétés des semi-conducteurs, pour comprendre les limites et les efficacités respectives de ces semi-conducteurs."
La modification des propriétés physiques d'un matériau modifie également ses propriétés électroniques. De petits changements dans la composition du matériau peuvent entraîner de grands changements dans les caractéristiques d'un matériau - dans certains cas, une légère altération atomique peut entraîner un changement de 1000 fois de la conductivité électrique.
Bien que les changements dans les propriétés des matériaux puissent être importants, les forces sous-jacentes exercées sur les atomes et les molécules et régissant leurs interactions sont généralement faibles et à courte portée (c'est-à-dire que les molécules et les atomes qui les composent doivent être proches les uns des autres). Pour comprendre les changements de propriétés, les chercheurs doivent calculer avec précision les interactions atomiques et moléculaires ainsi que les densités d'électrons et la façon dont ils sont transférés entre les molécules.
L'introduction d'atomes ou de molécules spécifiques dans un matériau peut modifier ses propriétés conductrices à un niveau hyperlocal. Cela permet à un transistor fabriqué à partir d'un matériau dopé de remplir une variété de rôles dans l'électronique, y compris le routage des courants pour effectuer des opérations basées sur des circuits complexes ou l'amplification du courant pour aider à produire du son dans un amplificateur de guitare ou une radio.
Les lois quantiques régissent les interactions interatomiques et intermoléculaires, essentiellement tenir le matériel ensemble, et structurer le monde tel que nous le connaissons. Dans le travail de l'équipe, ces interactions complexes doivent être calculées pour des interactions atomiques individuelles, y compris les interactions entre les molécules "hôtes" semi-conductrices et les molécules dopantes à plus grande échelle.
L'équipe utilise la théorie de la fonctionnelle de la densité (DFT), une méthode de calcul qui peut modéliser les densités et propriétés électroniques lors d'une interaction chimique, pour prédire efficacement la variété des interactions complexes. Il collabore ensuite avec des expérimentateurs de la TU Dresden et de l'Institute for Molecular Science d'Okazaki, Japon pour comparer ses simulations aux expériences de spectroscopie.
"La conductivité électrique peut provenir de nombreux dopants et est une propriété qui émerge sur une échelle de longueur beaucoup plus grande que les forces interatomiques, " a déclaré Ortmann. " La simulation de ce processus nécessite des modèles de transport plus sophistiqués, qui ne peut être implémenté que sur des architectures de calcul haute performance (HPC).
Pour tester son approche informatique, l'équipe a simulé des matériaux qui avaient déjà de bons ensembles de données expérimentales ainsi que des applications industrielles. Les chercheurs se sont d'abord concentrés sur le C60, également connu sous le nom de Buckminsterfullerene.
Le Buckminsterfullerene est utilisé dans plusieurs applications, y compris les cellules solaires. La structure de la molécule est similaire à celle d'un ballon de football - un arrangement sphérique d'atomes de carbone disposés en motifs pentagonaux et hexagonaux de la taille de moins d'un nanomètre. En outre, les chercheurs ont simulé la phtalocyanine de zinc (ZnPc), une autre molécule utilisée dans le photovoltaïque, mais contrairement au C60, a une forme plate et contient un atome métallique (zinc).
En tant que dopant, l'équipe a d'abord utilisé une molécule bien étudiée appelée 2-Cyc-DMBI (2-cyclohexyl-diméthylbenzimidazoline). Le 2-Cyc-DMBI est considéré comme un n-dopant, ce qui signifie qu'il peut fournir ses électrons excédentaires au semi-conducteur pour augmenter sa conductivité. Les N-dopants sont relativement rares, car peu de molécules sont « disposées » à céder un électron. Dans la plupart des cas, les molécules qui le font deviennent instables et se dégradent au cours des réactions chimiques, ce qui peut entraîner une panne de l'appareil électronique. Mais les dopants 2-Cyc-DMBI sont l'exception, car ils peuvent être suffisamment faiblement attractifs pour les électrons - leur permettant de se déplacer sur de longues distances - tout en restant stables après leur avoir fait don.
L'équipe a obtenu un bon accord entre ses simulations et les observations expérimentales des mêmes interactions molécule-dopant. Cela indique qu'ils peuvent s'appuyer sur la simulation pour guider les prédictions en ce qui concerne le processus de dopage des semi-conducteurs. Ils travaillent maintenant sur des molécules et des dopants plus complexes en utilisant les mêmes méthodes.
Malgré ces avancées, l'équipe reconnaît que les supercalculateurs de nouvelle génération tels que SuperMUC-NG, annoncés en décembre 2017 et devant être installés en 2018, aideront les chercheurs à élargir la portée de leurs simulations, conduisant à des gains d'efficacité toujours plus importants dans une variété d'applications électroniques.
« Nous devons pousser la précision de nos simulations au maximum, " a déclaré Ortmann. "Cela nous aiderait à étendre la gamme d'applicabilité et nous permettrait de simuler plus précisément un ensemble plus large de matériaux ou de plus grands systèmes de plus d'atomes."
Ortmann a également noté que si les systèmes de génération actuelle ont permis à l'équipe de mieux comprendre des situations spécifiques et de prouver son concept, il y a encore de la place pour s'améliorer. « Nous sommes souvent limités par la mémoire système ou la puissance du processeur, " Il a dit. " La taille du système et la précision de la simulation sont essentiellement en compétition pour la puissance de calcul, c'est pourquoi il est important d'avoir accès à de meilleurs supercalculateurs. Les supercalculateurs sont parfaitement adaptés pour apporter des réponses à ces problèmes dans un délai réaliste."