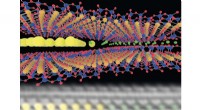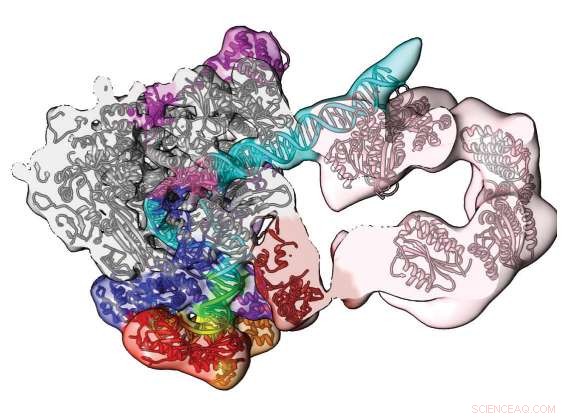
Le calcul intensif et la microscopie cryoélectronique révèlent cette section du complexe de pré-initiation humain. La carte et le modèle de densité de conformation ouverte montrent le chemin de l'ADN (bleu/vert) et son engagement par le composant du facteur de transcription TFIIH (rose). Réimprimé avec la permission de Macmillan Publishers Ltd :He, Y. et al. Visualisation à résolution quasi atomique de l'ouverture du promoteur de la transcription humaine. La nature 533, 359-365 (2016).
Cela ressemble à quelque chose des Borgs dans Star Trek. Des robots de taille nanométrique s'auto-assemblent pour former des machines biologiques qui font le travail qui permet de rester en vie. Et pourtant, quelque chose comme ça se passe vraiment.
Chaque cellule de notre corps - qu'elles soient de chair et de sang, cerveau et tout le reste - a un ADN identique, l'escalier tordu d'acides nucléiques codés de manière unique à chaque organisme. Des assemblages complexes qui ressemblent à des machines moléculaires prélèvent des morceaux d'ADN appelés gènes et fabriquent une cellule cérébrale en cas de besoin, à la place de, dire, une cellule osseuse. Ces machines moléculaires sont si complexes, pourtant si petit, que les scientifiques d'aujourd'hui commencent tout juste à comprendre leur structure et leur fonction à l'aide des derniers microscopes et superordinateurs. Les machines moléculaires biologiques pourraient jeter les bases du développement de traitements contre des maladies comme le cancer. Comme on peut voir petit, et que trouvera-t-on ?
La microscopie cryoélectronique combinée à des simulations sur superordinateur ont créé le meilleur modèle à ce jour, avec des détails proches du niveau atomique, d'une machine moléculaire vitale, le complexe de pré-initiation humaine (PIC). Une équipe scientifique de la Northwestern University, Laboratoire national de Berkeley, Université d'État de Géorgie, et UC Berkeley ont publié leurs résultats sur le PIC en mai 2016 dans la revue La nature .
"Pour la première fois, les structures ont été détaillées des groupes complexes de molécules qui ouvrent l'ADN humain, " a déclaré le co-auteur de l'étude Ivaylo Ivanov, professeur agrégé de chimie à la Georgia State University. Ivanov a dirigé le travail informatique qui a modélisé les atomes des différentes protéines qui agissent comme les rouages de la machine moléculaire PIC.
Le PIC trouve des gènes associés à la fabrication d'une protéine spécifique, tel qu'un anticorps ou une enzyme. Là, le PIC sépare les deux brins d'ADN et alimente le brin codant pour l'enzyme cheval de bataille ARN polymérase II. Cela commence la transcription, où les bits d'ADN sont copiés par l'ARN polymérase II en un seul brin d'ARN messager. L'ARN se dirige vers des «usines à protéines» dans la cellule appelées ribosomes qui les prennent comme commandes pour quelle protéine fabriquer. Si l'ADN est comme le plan d'une nouvelle maison, Les ARN sont des instructions aux « contractants » au poste de travail du ribosome. Les protéines fabriquées sont comme les ongles, bois, plâtre, et à peu près tout le reste dans la maison.
L'expérience a commencé avec des images minutieusement prises de PIC. Ils ont été réalisés par un groupe dirigé par la co-auteur de l'étude Eva Nogales, professeur au Département de biologie moléculaire et cellulaire de l'UC Berkeley et également scientifique principal de la faculté au Lawrence Berkeley National Laboratory et chercheur médical Howard Hughes.
Le groupe de Nogales a utilisé la cryomicroscopie électronique (cryo-EM), une étoile montante des techniques de laboratoire. Ils ont congelé par cryogénie du PIC humain lié à l'ADN. La congélation l'a maintenu dans une substance chimiquement active, environnement proche de la nature. Ensuite, ils l'ont zappé avec des faisceaux d'électrons. Grâce aux récents progrès de la technologie des détecteurs d'électrons directs, cryo-EM peut désormais imager à une résolution proche de l'atome de grandes structures biologiques complexes qui se sont avérées trop difficiles à cristalliser. La technique incontournable, Cristallographie aux rayons X, nécessite des spécimens cristallisés, et cryo-EM évite cette étape difficile.
Plus de 1,4 million d'images fixes cryo-EM de PIC ont été traitées à l'aide de superordinateurs du National Energy Research for Scientific Computing Center pour filtrer le bruit de fond et reconstruire des cartes de densité tridimensionnelles qui montrent des détails sous la forme de la molécule qui n'avaient jamais été Déjà vu.
"Cryo-EM connaît une grande expansion, tout comme tous les logiciels informatiques utilisés pour générer à la fois les cartes de densité et aussi pour les interpréter comme nous l'avons fait dans cette étude, " a déclaré Nogales. " Cela nous permet d'obtenir une résolution plus élevée de plus de structures dans différents états afin que nous puissions décrire non seulement une image de leur apparence, mais plusieurs photos montrant comment ils se déplacent. Nous ne voyons pas de continuum, mais nous voyons des instantanés à travers le processus d'action."
Les scientifiques de l'étude ont ensuite construit un modèle précis qui donne un sens physique aux cartes de densité du PIC à l'aide de XSEDE, l'environnement de découverte scientifique et technique eXtream, financé par la National Science Foundation. XSEDE permet aux scientifiques de partager de manière interactive des ressources informatiques, données et expertise via un système virtuel unique. L'équipe d'Ivaylo Ivanov a exécuté plus de quatre millions d'heures de simulation sur le supercalculateur Stampede du Texas Advanced Computing Center pour modéliser des machines moléculaires complexes, y compris ceux de cette étude. Le travail plus large d'Ivanov sur les machines moléculaires comprend également une allocation XSEDE de 1,7 million d'heures de base sur le supercalculateur Comet du San Diego Supercomputing Center.
"J'utilise les ressources XSEDE depuis plus de 12 ans maintenant, " a déclaré Ivanov. " Sans la disponibilité des ressources XSEDE, toutes nos recherches auraient été beaucoup plus limitées en termes de systèmes que nous pouvons aborder. Pour nous, XSEDE a été absolument essentiel."
L'objectif de tout cet effort de calcul est de produire des modèles atomiques qui racontent toute l'histoire de la structure et de la fonction du complexe protéique des molécules. Pour y arriver, l'équipe d'Ivanov a pris les douze composants de l'assemblage PIC et a créé des modèles d'homologie pour chaque composant qui représentaient leurs séquences d'acides aminés et leur relation avec des structures 3-D de protéines connues similaires.
Ensuite, ils ont approximé les densités expérimentales que l'équipe de Nogales a trouvées sur une grille. « Nous pouvons utiliser une méthode appelée ajustement flexible de la dynamique moléculaire, " expliqua Ivanov, "où vous exécutez essentiellement une simulation de dynamique moléculaire. Et vous utilisez la densité expérimentale pour biaiser les atomes dans la simulation de dynamique moléculaire pour se déplacer dans les régions les plus denses de la carte EM. C'est le processus d'ajustement flexible à la carte EM. "
Ils ont affiné le modèle avec le package de raffinement cristallographique Phoenix. "C'est une technique complémentaire qui nous permet de positionner les chaînes latérales et d'améliorer le modèle afin que nous puissions capturer tous les détails qui sont présents dans la carte de densité, ", a déclaré Ivanov.
XSEDE était "absolument nécessaire" pour cette modélisation, dit Ivanov. "Lorsque nous incluons de l'eau et des contre-ions en plus du complexe PIC dans une boîte de simulation de dynamique moléculaire, nous obtenons la taille du système de simulation de plus d'un million d'atomes. On ne peut pas faire ça sur un poste de travail ou même sur un cluster modeste. Pour cela, nous avons vraiment besoin d'aller à un millier de cœurs. Dans ce cas, nous sommes allés jusqu'à deux mille quarante-huit cœurs. Et pour cela, nous avions besoin d'accéder à Stampede, ", a déclaré Ivanov.
L'une des connaissances acquises dans l'étude est un modèle de travail de la façon dont le PIC ouvre la double hélice d'ADN par ailleurs stable pour la transcription. Nogales a expliqué qu'on pouvait imaginer une corde faite de deux fils enroulés l'un autour de l'autre. Tenez une extrémité très fermement. Saisissez l'autre et tournez-le dans le sens inverse de l'enfilage pour démêler le cordon. C'est essentiellement ainsi que procèdent les machines vivantes qui nous maintiennent en vie.
"L'ADN doit être ouvert et déplacé dans le site actif de la polymérase pour coder pour le premier nucléotide d'ARN, " a expliqué Nogales. " Le complexe de pré-initiation maintient les deux brins de l'ADN très étroitement ensemble à une extrémité, afin qu'ils ne puissent pas bouger et qu'ils ne puissent pas s'ouvrir. De l'autre côté du PIC il y a une machine qui utilise de l'énergie pour pousser l'ADN, en le tordant dans le sens opposé à l'enfilage des deux brins. Et quand cela arrive, entre les deux côtés, les brins s'ouvriront, " dit Nogales.
Cette étude a résolu la structure de cette machine moléculaire qui agit comme les doigts de torsion, le composant du facteur de transcription TFIIH. « TFIIH dispose d'une sous-unité translocase, dont le rôle est de pousser simultanément l'ADN vers le site actif de la polymérase et de dérouler l'ADN. Par la poussée et le déroulement combinés, effectivement vous séparez les deux brins de l'ADN, ", a déclaré Ivanov.
Les deux scientifiques ont déclaré qu'ils commençaient tout juste à comprendre la transcription au niveau atomique, crucial pour l'expression des gènes et finalement la maladie. « De nombreux états pathologiques surviennent parce qu'il y a des erreurs dans la quantité de lecture d'un certain gène et la présence d'une certaine protéine avec une certaine activité dans la cellule, " Nogales a déclaré. "Ces états pathologiques pourraient être dus à une production excessive de la protéine, ou au contraire pas assez. Il est très important de comprendre le processus moléculaire qui régule cette production afin que nous puissions comprendre l'état de la maladie."
"Ce travail illustre bien deux principes généraux qui guideront la science dans les prochaines années, " a commenté Peter Preusch, responsable de programme auprès des National Institutes of Health (NIH). "L'une est l'application de méthodes hybrides - des combinaisons de méthodes biophysiques, notamment la cristallographie aux rayons X et la cryoEM, ainsi que des méthodes de calcul à grande échelle pour intégrer des informations sur des complexes moléculaires plus importants. Deux, il est nécessaire que la science d'équipe fasse appel à l'expertise de plusieurs chercheurs pour résoudre des problèmes qui ne peuvent être résolus par un seul laboratoire travaillant seul. » Peter Preusch est le chef de la branche de biophysique, Division de biologie cellulaire et biophysique, Institut national des sciences médicales générales, NIH.
Bien que ce travail fondamental ne produise pas directement de remèdes, il jette les bases pour aider à les développer à l'avenir, dit Ivanov. « Pour comprendre la maladie, nous devons comprendre comment ces complexes fonctionnent en premier lieu… Une collaboration entre les modélisateurs informatiques et les biologistes structurels expérimentaux pourrait être très fructueuse à l'avenir. "
L'étude Nature Articles de mai 2016 (DOI :10.1038/nature17970), "Visualisation à résolution quasi atomique de l'ouverture du promoteur de transcription humaine, " a été écrit par Yuan He, Lawrence Berkeley National Laboratory et maintenant à l'Université Northwestern; Chunli Yan et Ivaylo Ivanov, Université d'État de Géorgie ; Jie Fang, Carla Inouye, Robert Tjian, Eva Nogales, UC Berkeley. Le financement provient de l'Institut national des sciences médicales générales (NIH) et de la National Science Foundation.