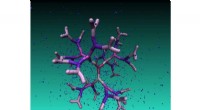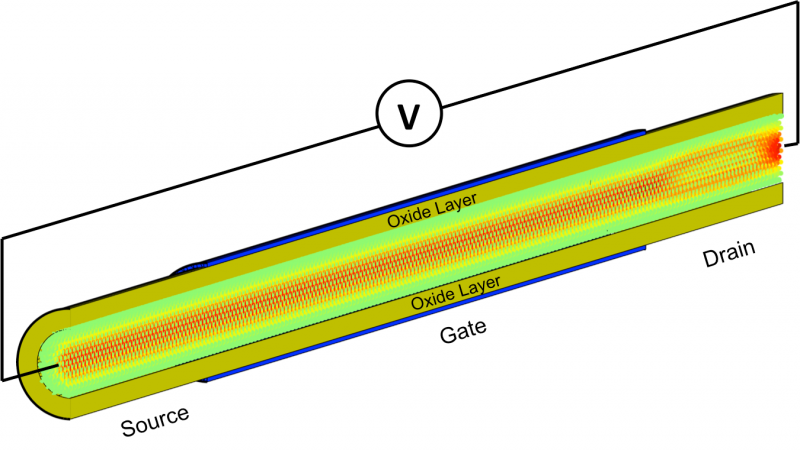
Distribution spatiale du courant d'électrons circulant à travers un transistor à effet de champ à nanofils à grille en silicium composé de 55, 488 atomes. Une tension (V) est appliquée à la structure. La moitié de la couche de coiffage d'oxyde est retirée pour éclairer l'intérieur du transistor où les atomes sont colorés en fonction du courant qu'ils transportent :le vert signifie pas de courant, tandis que le rouge indique une concentration élevée.
Les chercheurs de l'ETH Zurich utilisent le superordinateur le plus rapide des États-Unis pour réaliser d'énormes progrès dans la compréhension des plus petits appareils électroniques.
L'équipe, dirigé par Mathieu Luisier, se concentre sur le développement de la première ligne de la recherche en électronique - simuler et mieux comprendre les composants à l'échelle nanométrique tels que les transistors ou les électrodes de batterie dont les régions actives peuvent être de l'ordre d'un milliardième de mètre, ou à peu près aussi longtemps que vos ongles poussent en une seconde.
Bien que les échelles des objets étudiés soient petites, l'équipe a fait de grands progrès vers des codes de calcul plus efficaces. Ses recherches ont été sélectionnées comme finaliste pour le prix Gordon Bell de l'Association of Computing Machinery de cette année, l'un des prix les plus prestigieux du calcul intensif.
La candidature de l'équipe est le résultat d'une recherche menée sur le supercalculateur Cray XK7 Titan de l'Oak Ridge Leadership Computing Facility. L'OLCF est une installation d'utilisateurs de l'Office of Science du département de l'Énergie des États-Unis située au Oak Ridge National Laboratory.
Ordinateurs portables, les téléphones portables et autres appareils électroniques deviennent moins chers et plus accessibles tout en devenant de plus en plus sophistiqués. Ces progrès sont en grande partie dus aux dimensions de plus en plus réduites de leurs composants électroniques.
Cependant, développer du matériel de nouvelle génération nécessite désormais que les scientifiques et les ingénieurs comprennent les interactions des matériaux à des échelles de temps et de taille extrêmement petites, amener les chercheurs à augmenter l'expérience par la simulation.
"Notre objectif est d'étudier des dispositifs à l'échelle nanométrique, tels que les nanotransistors, piles ou une variété d'autres nouveaux appareils tels que les mémoires d'ordinateur, interrupteurs optiques ou diodes électroluminescentes au niveau atomique, " a déclaré Luisier. " Si vous voulez rendre ces simulations précises et vraiment prédictives, vous devez utiliser ce qu'on appelle ab initio, ou des premiers principes, méthodes de simulation."
Essentiellement, Les simulations ab initio permettent aux chercheurs de modéliser n'importe quel système atomique à partir de zéro sans avoir besoin de paramètres de matériaux pré-calibrés. Bien sûr, atteindre un tel niveau de précision n'est pas gratuit. Le prix est mille fois supérieur à la complexité de calcul par rapport à, par exemple, approches semi-empiriques qui utilisent les données d'expériences pour simplifier le calcul.
Les chercheurs qui étudient la nanoélectronique doivent donc généralement faire un compromis entre la simulation d'une taille de système réaliste (au moins 10, 000 atomes) et en utilisant des méthodes ab initio très précises.
A ce point, bien que, la plupart des progiciels ab initio se concentrent sur le calcul des propriétés des matériaux tels que les structures cristallines et électroniques, vibrations du réseau, ou des diagrammes de phases et ne tiennent pas compte des conditions réelles de fonctionnement - sous l'application d'une tension externe, un courant d'électrons commence à circuler à travers les nanostructures actives. Ces phénomènes de transport sont très gourmands en calcul et nécessitent une approche de modélisation dédiée.
Luisier et son équipe, donc, a développé une méthode pour faire des simulations de transport ab initio qui sont suffisamment grandes pour étudier des nanostructures avec des tailles pertinentes pour l'industrie et les groupes expérimentaux. Ils avaient juste besoin de la bonne machine pour le tester.
Deux codes partenaires, un objectif
Les circuits intégrés d'aujourd'hui sont composés de plusieurs milliards de transistors étroitement entassés sur une surface qui ne dépasse pas quelques centimètres carrés. Avec la nanoélectronique, on pourrait adapter des milliers de nanotransistors actuellement fabriqués dans la largeur d'un cheveu humain. Ces systèmes sont si petits que les chercheurs doivent recourir à la théorie quantique pour comprendre leurs propriétés.
L'équipe utilise deux progiciels différents pour accomplir cette tâche. Le code communautaire CP2K, développé et maintenu par le professeur de l'ETH Zurich Joost VandeVondele, fournit la description ab initio des nanostructures, tandis que le code OMEN du groupe de Luisier effectue les simulations de transport quantique basées sur les entrées de CP2K. En combinant CP2K et OMEN, l'équipe peut obtenir une perspective unique « matériau + appareil » des systèmes atomiques.
Luisier a expliqué qu'il y a deux défis principaux pour simuler le transport à travers des composants nanoélectroniques. D'abord, les chercheurs doivent calculer ce qu'ils appellent des conditions aux limites ouvertes qui couplent la simulation avec son environnement environnant et permettent les flux de courant. Dans un deuxième temps, ils doivent intégrer les blocs frontières créés dans l'hamiltonien, une matrice qui contient l'ensemble des interactions interatomiques caractérisant le dispositif, et enfin ils doivent résoudre le système linéaire clairsemé d'équations résultant. En utilisant cette approche, les simulations de pointe typiques sur le terrain peuvent modéliser avec précision environ 1, 000 atomes.
Avec l'émergence des supercalculateurs hybrides, l'équipe s'est rendu compte qu'elle avait besoin d'une nouvelle approche de simulation capable d'exploiter le potentiel des processeurs et des accélérateurs GPU. En gardant cette idée en tête, deux doctorants dans le groupe de Luisier, Sascha Bruck et Mauro Calderara, a mis en place un schéma original permettant à l'équipe de calculer simultanément les conditions aux limites ouvertes sur les CPU et de créer la matrice hamiltonienne appropriée sur les GPU avant une courte phase de post-traitement, puis combiner les deux résultats. Ce tour de force a non seulement permis de décharger le travail sur les GPU, mais a également attaqué le problème sur deux fronts en même temps, réduisant considérablement le temps de simulation.
"Ce qui nous a permis d'aller beaucoup plus vite et de traiter de très grandes structures d'appareils, c'est que nous avons trouvé un moyen d'effectuer efficacement la plupart du travail, résoudre le système linéaire, sur les nœuds de calcul de Titan, utilisant des GPU extrêmement rapides, tout en gardant les CPU occupés à calculer les conditions aux limites en même temps, " dit Luisier.
L'équipe a d'abord testé sa méthode sur la machine Piz Daint du Centre national suisse de calcul intensif, croissance de la simulation à partir de 1, 000 atomes à 15, 000. Pour Luisier, c'était extrêmement encourageant, mais il croyait que l'équipe pouvait faire plus.
Après ces premières courses réussies, l'équipe a reçu du temps sur Titan dans le cadre du programme discrétionnaire du directeur. Déménagement du Piz Daint, avec ses 5, Plus de 000 nœuds de calcul, à Titan - avec plus de 18 ans, 000 nœuds - a permis à l'équipe d'effectuer une simulation avec 50, 000 atomes, battre facilement la référence précédente. Luisier a également noté qu'atteindre un 50, La simulation à 000 atomes n'a même pas utilisé toute la puissance de supercalcul de Titan, ce qui signifie que les simulations plus importantes ne sont pas seulement théoriques, mais probablement, dans le futur proche.
En trouvant une méthode pour faire des calculs de transport quantique ab initio sur un si grand système, l'équipe est la première à réaliser des simulations pouvant correspondre à des expérimentations sur le terrain, potentiellement aider à faire avancer la recherche et le développement pour les appareils électroniques de la prochaine génération.
"Si vous n'en avez que 1, 000 atomes, vous ne pouvez pas vraiment simuler un vrai appareil, " Luisier a dit. "Cela nécessiterait de simuler environ 10 fois plus d'entre eux. Avec la nouvelle méthode, nous pouvons vraiment modéliser quelque chose qui ressemble à un transistor ou à une unité de stockage au niveau ab initio. Et les nanofils que nous avons étudiés ont déjà été fabriqués il y a environ 10 ans, lorsque les expérimentateurs n'étaient pas aussi avancés qu'aujourd'hui dans la production de petites structures. Ainsi, le maximum de ce que nous pouvons maintenant simuler va au-delà des plus petites structures que les gens peuvent réellement fabriquer en laboratoire aujourd'hui. »
Bien que la performance soutenue des codes soit impressionnante -15 pétaflops, ou 15 quadrillions de calculs par seconde - Luisier a souligné que ces simulations n'ont pas été effectuées pour établir de nouvelles références de performances de calcul dans le domaine, mais plutôt de poursuivre les recherches.
"C'est vraiment un code de production, un code utilisé au quotidien, " a déclaré Luisier. "Ce qui ressort de ces essais n'est pas seulement FLOPS sur un ordinateur - ces résultats sont utilisés en collaboration avec des expérimentateurs à l'ETH Zürich et à l'étranger. Il y a quelques groupes très intéressés par les résultats car ils peuvent expliquer ce que ces groupes observent dans leurs dispositifs expérimentaux - non seulement dans les nanotransistors mais aussi dans les composants électroluminescents ou les cellules solaires à points quantiques, pour ne citer que quelques exemples."