Tout comme les humains qui les ont créés, les ordinateurs trouvent la physique difficile, mais la mécanique quantique encore plus. Mais une nouvelle technique créée par trois scientifiques de l'Université de Chicago permet aux ordinateurs de simuler certains effets mécaniques quantiques difficiles dans des matériaux électroniques complexes avec beaucoup moins d'effort.
En rendant ces simulations plus précises et efficaces, les scientifiques espèrent que cette technique pourrait aider à découvrir de nouvelles molécules et de nouveaux matériaux, tels que de nouveaux types de cellules solaires ou d'ordinateurs quantiques.
"Cette avancée recèle un immense potentiel pour approfondir notre compréhension des phénomènes moléculaires, avec des implications significatives pour la chimie, la science des matériaux et les domaines connexes", a déclaré le scientifique Daniel Gibney, titulaire d'un doctorat à l'Université de Chicago. étudiant en chimie et premier auteur de l'article, publié le 14 décembre dans Physical Review Letters .
Une feuille ou un panneau solaire semble lisse et simple de l'extérieur, mais zoomez jusqu'au niveau moléculaire et vous verrez une danse extrêmement complexe d'électrons et de molécules.
Afin de réaliser de nouveaux progrès dans les domaines du développement durable, de l’industrie manufacturière, de l’agriculture et de nombreux autres domaines, les scientifiques modélisent le comportement de ces interactions chimiques et moléculaires. Cela permet de révéler de nouvelles possibilités de conception pour l'avenir, depuis les nouvelles façons de séquestrer le dioxyde de carbone jusqu'aux nouveaux types de bits quantiques.
De nombreux progrès ont été réalisés au cours des dernières décennies, mais l'un des domaines qui reste obstinément difficile à simuler est celui où les molécules commencent à afficher des comportements mécaniques quantiques complexes que les scientifiques appellent une forte corrélation.
Le problème est qu’une fois que les électrons commencent à manifester leurs effets les plus quantiques – comme s’ils s’ « enchevêtrent » – les calculs nécessitent instantanément beaucoup plus de puissance de calcul. Même les superordinateurs ont du mal à gérer les implications.
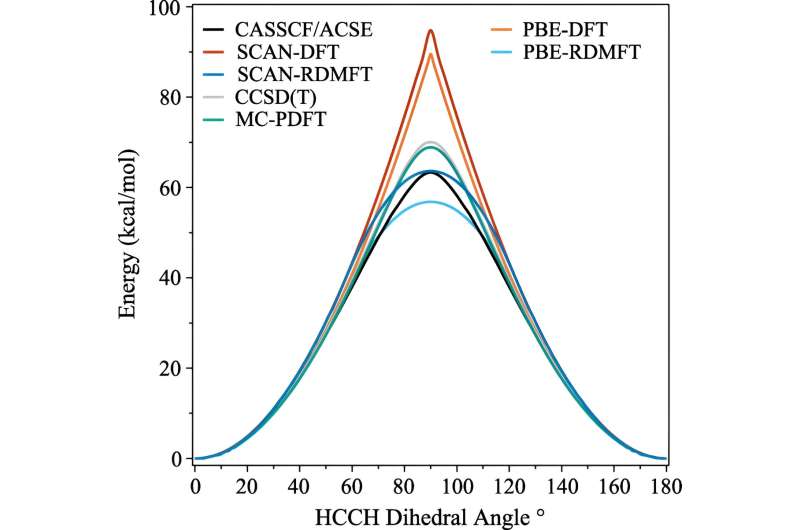
L’un des calculs couramment utilisés est appelé théorie fonctionnelle de la densité. "Il s'agit fondamentalement de la technique la plus omniprésente pour prédire la structure électronique, mais il s'agit essentiellement d'une approximation où tous les électrons sont traités en fonction d'un électron", a expliqué David Mazziotti, professeur de chimie et auteur principal de l'étude.
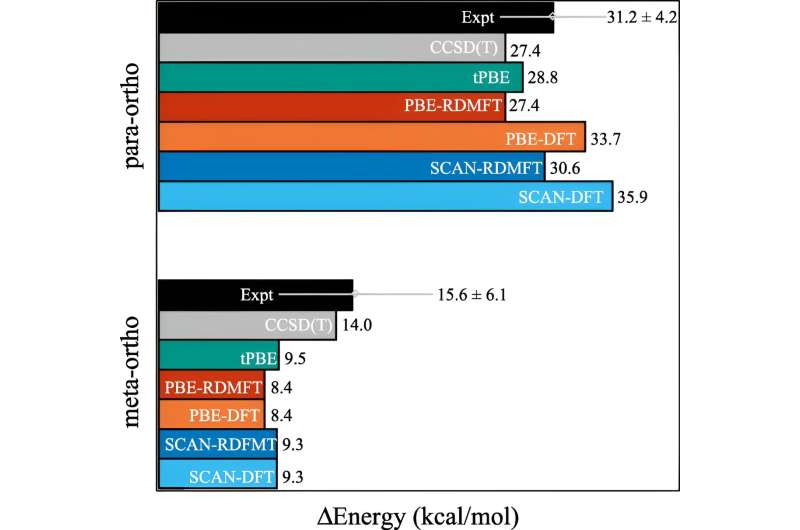
Pour de nombreux calculs, une approximation fait l’affaire. Mais cela commence à s’effondrer à mesure que le comportement des électrons devient plus corrélé, comme cela se produit lorsque la mécanique quantique commence à entrer en jeu. En mécanique quantique, ces électrons peuvent se trouver simultanément à plusieurs endroits, ou orbitales. Cela contrecarre non seulement le cerveau humain, mais aussi la théorie de la densité fonctionnelle.
"Et c'est un problème important, car bon nombre des problèmes qui nous préoccupent au 21e siècle, comme les nouvelles molécules et les nouveaux matériaux pour les énergies renouvelables et la durabilité, nous obligent à exploiter la nature quantique des matériaux", a déclaré Mazziotti.
Mazziotti, Gibney et le troisième auteur Jan-Niklas Boyn ont découvert qu'ils pouvaient ajouter une correction universelle à la théorie fonctionnelle de la densité qui permet aux électrons de s'emmêler simultanément sur plusieurs orbitales.
"Cela permet aux orbitales du calcul d'être non seulement entièrement remplies ou entièrement vides, mais également n'importe où entre les deux", a déclaré Mazziotti. "Nous arrivons à une image à un électron qui est encore capable de capturer le comportement qui résulte des effets électroniques corrélés à plusieurs corps."
En prime, selon les scientifiques, le code peut être ajouté aux algorithmes existants sans avoir à réécrire ce code. "Fondamentalement, la correction intervient chaque fois que cela est nécessaire, mais n'interfère pas avec le reste du code dans le cas contraire", a déclaré Gibney.
Il est également universel :il peut être ajouté au code simulant de nombreux types de comportement électronique, qu'il s'agisse de panneaux solaires photovoltaïques, de séquestration du carbone ou de matériaux supraconducteurs, voire même de la biologie.
Par exemple, a expliqué Boyn, une application pourrait consister à comprendre la chimie qui se produit à l'aide d'enzymes contenant des atomes métalliques, appelées métalloenzymes.
"Il existe une pléthore de métalloenzymes responsables d'une grande partie de la chimie de vos cellules, par exemple, mais elles sont notoirement difficiles à décrire avec les modèles actuels", a-t-il déclaré. "Cette théorie pourrait dans un avenir proche nous permettre d'aborder cette chimie d'une manière qui est impossible à l'heure actuelle."
Plus d'informations : Daniel Gibney et al, Généralisation universelle de la théorie fonctionnelle de la densité pour la corrélation statique, Physical Review Letters (2023). DOI : 10.1103/PhysRevLett.131.243003
Informations sur le journal : Lettres d'examen physique
Fourni par l'Université de Chicago