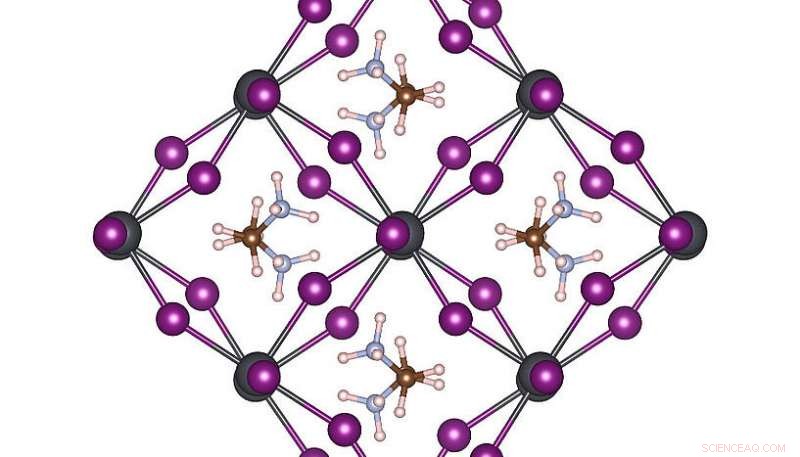
Structure atomique à haute symétrie de MAPbI3 à température ambiante. Crédit :Menno Bokdam/Université de Vienne
A l'échelle atomique, les matériaux peuvent montrer une riche palette de comportements dynamiques, qui affecte directement les propriétés physiques de ces matériaux. Pendant de nombreuses années, c'était un rêve de décrire cette dynamique dans des matériaux complexes à différentes températures à l'aide de simulations informatiques. Des physiciens de l'Université de Vienne ont développé une méthode d'apprentissage automatique à la volée qui permet de tels calculs grâce à une intégration directe dans le Vienna Ab-initio Simulation Package (VASP) basé sur la mécanique quantique. La polyvalence de la méthode d'auto-apprentissage est démontrée par de nouvelles découvertes, publié dans la revue Lettres d'examen physique , sur les transitions de phase des pérovskites hybrides. Ces pérovskites sont d'un grand intérêt scientifique en raison de leur potentiel dans la récupération d'énergie solaire et d'autres applications.
À température ambiante, tous les matériaux se déplacent constamment à l'échelle atomique. Même la roche solide est constituée d'atomes qui oscillent. Les propriétés physiques des matériaux sont directement liées à la disposition des atomes dans le, ainsi appelé, réseau cristallin. Selon la température ou la pression, cette disposition peut changer, affectant ainsi les propriétés des matériaux. On peut penser au diamant, qui est transparent et dur en raison de l'arrangement périodique des atomes de carbone dans le cristal de diamant. Les mêmes atomes, disposé différemment, résultats en noir, graphite cassant. Il était déjà possible de calculer avec précision les coordonnées des atomes dans des matériaux simples à différentes températures avec des simulations de mécanique moléculaire quantique (MD). Cependant, de tels calculs sont coûteux en temps de calcul et limitent les applications pratiques à quelques centaines d'atomes et un temps de simulation limité.
Des physiciens du groupe Computational Materials Physics de l'Université de Vienne ont développé une nouvelle approche qui surmonte ces limitations et rend possible des simulations de matériaux complexes pour de futures applications énergétiques. Ceci est réalisé en développant un algorithme d'auto-apprentissage efficace et robuste basé sur les données et, le plus important, en intégrant cet algorithme directement dans le Vienna Ab-initio Simulation Package (VASP). Dans la nouvelle approche, la "machine" peut ramasser, tout seul, les ingrédients essentiels pour une description plus simple du modèle des atomes en interaction lors des simulations MD. Déjà après avoir calculé quelques centaines de pas de temps, la machine peut prédire avec suffisamment de précision les positions des atomes dans le pas de temps consécutif. La machine est également capable de faire une estimation de sa précision pour les étapes consécutives. Si l'erreur est trop élevée, la machine change de vitesse et effectue le très précis, Mais cher, calculs DM. Plus le temps de simulation passe, plus la machine apprend et plus elle devient précise. De cette façon, de moins en moins de calculs MD sont nécessaires, ce qui conduit finalement à la situation où tous les pas de temps sont effectués par la machine. De plus, la capacité d'auto-apprentissage à la volée réduit le besoin d'intervention humaine requis par d'autres méthodes d'apprentissage automatique existantes.
Pour démontrer la puissance de cette nouvelle méthode, les chercheurs l'ont appliqué pour étudier les transitions entre différentes structures atomiques du MAPbI
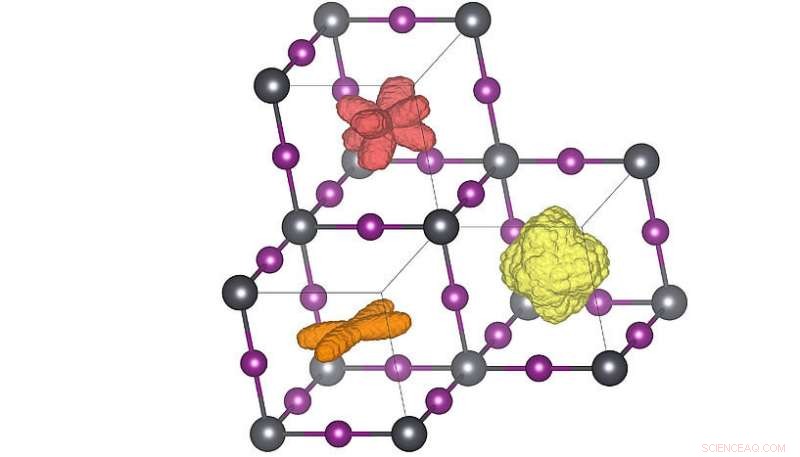
Distributions tridimensionnelles de l'orientation de la molécule dans les trois phases cristallines différentes. Lorsque la température est élevée (orange → rouge → jaune), les molécules peuvent atteindre plus d'orientations. La répartition rouge correspond à la structure de la température ambiante. Crédit :Menno Bokdam/Université de Vienne