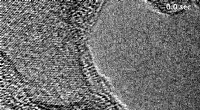
Modèle structural de la fibrille peptidique d'Alzheimer Amyloïde A-beta 1-42 dérivée d'une structure expérimentale (PDB :2MXU). Les agrégats fibrillaires agissent comme des toxines cellulaires au début et à la progression de la maladie d'Alzheimer. Crédit :Emmanuel Peter
Protéines, les chevaux de bataille omniprésents de la biochimie, sont d'énormes molécules dont la fonction dépend de la façon dont elles se replient dans des structures complexes. Pour comprendre le fonctionnement de ces molécules, les chercheurs utilisent la modélisation informatique pour calculer le repliement des protéines.
Maintenant, un nouvel algorithme peut accélérer ces simulations vitales, leur permettant de modéliser des phénomènes qui étaient auparavant hors de portée. Les résultats peuvent éventuellement aider les scientifiques à mieux comprendre et traiter des maladies comme la maladie d'Alzheimer, dit Emmanuel Pierre, chimiste à l'Université de Ratisbonne. Son travail sur la nouvelle technique est décrit cette semaine dans Le Journal de Physique Chimique .
Simulations conventionnelles, en utilisant la dynamique moléculaire et les méthodes de Monte Carlo, ont réussi dans l'ensemble à modéliser des molécules biologiques comme des protéines. Pour déterminer comment les protéines se replient, la simulation recherche des configurations qui correspondent à des états d'énergie de plus en plus faibles. Finalement, il trouve l'état d'énergie le plus bas, ce qui donne un pli stable. Mais pendant que la simulation cherche, il peut rencontrer une configuration avec une énergie légèrement supérieure, qui forme une barrière qui entrave l'algorithme.
En raison de ces ralentissements, les méthodes conventionnelles ne peuvent simuler que des comportements moléculaires se produisant sur des échelles de temps courtes de quelques centaines de microsecondes. De nombreux phénomènes, tels que certains replis protéiques ou un médicament se liant à une cible potentielle, se dérouler en quelques secondes, minutes voire des jours. Simuler des échelles de temps aussi longues prendrait trop de temps de calcul avec des approches conventionnelles.
Pour accélérer les simulations, les chercheurs peuvent injecter de l'énergie dans le système, qui pousse le modèle au-dessus de toutes les barrières énergétiques. Mais l'un des plus grands défis de ces méthodes est de définir les coordonnées qui décrivent le système - qui, par exemple, peut être la longueur entre les atomes dans la molécule, et les angles entre les liaisons. Traditionnellement, les chercheurs définissent les coordonnées avant de commencer la simulation. Chaque pas de temps le long de chaque coordonnée dépend de l'étape précédente. Mais cette dépendance peut biaiser la simulation.
Le nouvel algorithme de Peter évite ce biais. Il a trouvé un système de coordonnées généralisé dans lequel chaque pas de temps ne repose pas sur le pas précédent. "Seulement quelques paramètres sont nécessaires, et aucune intuition humaine n'est requise, qui peut potentiellement biaiser le résultat de la simulation, " il a dit.
Pour tester le nouvel algorithme, Peter l'a utilisé pour modeler l'eau, un peptide appelé dialanine, le repliement d'un autre peptide appelé TrpCage, et l'agglutination de bêta-amyloïde 25-35, qui sont des fragments de protéines associés à la maladie d'Alzheimer. Dans chaque cas, sa technique rapporte avoir accéléré les simulations. Et les simulations de bêta-amyloïde pourraient aider à expliquer pourquoi la maladie d'Alzheimer a été difficile à traiter.
Dans la maladie d'Alzheimer, les fragments de protéine amyloïde-bêta s'agrègent ensemble, formation de plaque dure qui s'accumule entre les neurones et les perturbe. La bêta-amyloïde est également une toxine, entraînant la mort des cellules neuronales et la dégénérescence de la fonction neuronale. Les nouvelles simulations suggèrent que le bêta-amyloïde peut assumer une gamme de structures. Cette flexibilité structurelle pourrait expliquer pourquoi certains médicaments qui tentent d'inhiber l'agrégation n'ont pas réussi, dit Pierre. Lorsque ces médicaments se lient à la bêta-amyloïde, le bêta-amyloïde change juste de forme, lui permettant de continuer à s'agglutiner. Le médicament s'incorpore à l'agrégat et à la plaque.
Ce type de flexibilité structurelle, appelée entropie de conformation, est également une caractéristique clé dans d'autres peptides qui forment des plaques toxiques dans des maladies telles que la maladie de Huntington, Diabète de type 2, et la maladie de Parkinson. Le nouvel algorithme pourrait donc être utile pour comprendre également ces autres maladies.