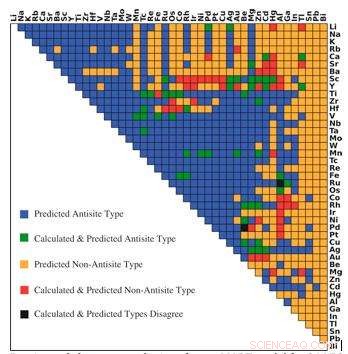
Prédictions de type de défaut dominant du modèle r-MART pour 946 intermétalliques de type B2. Les couleurs indiquent la relation entre la prédiction et les calculs, comme indiqué dans la légende. Crédit :Bharat Medasani, Laboratoire de Berkeley / PNNL
Pour la première fois, des chercheurs du Lawrence Berkeley National Laboratory (Berkeley Lab) ont construit et formé des algorithmes d'apprentissage automatique pour prédire le comportement des défauts dans certains composés intermétalliques avec une grande précision. Cette méthode accélérera la recherche de nouveaux alliages avancés et de nouveaux matériaux légers pour des applications allant de l'automobile à l'aérospatiale et bien plus encore.
Leurs résultats ont été publiés dans le numéro de décembre 2016 de Matériaux de calcul de la nature .
Les matériaux ne sont jamais chimiquement purs et structurellement irréprochables. Ils contiennent presque toujours des défauts, qui jouent un rôle important dans la dictée des propriétés. Ces défauts peuvent apparaître comme des vacances, qui sont essentiellement des « trous » dans la structure cristalline de la substance, ou défauts antisites, qui sont essentiellement des atomes placés sur le mauvais site cristallin. La compréhension de ces défauts ponctuels est cruciale pour les scientifiques qui conçoivent des matériaux, car ils peuvent avoir un effet dramatique sur la stabilité et la résistance structurelles à long terme.
Traditionnellement, les chercheurs ont utilisé une méthode de mécanique quantique informatique connue sous le nom de calculs fonctionnels de densité pour prédire quels types de défauts peuvent se former dans une structure donnée et comment ils affectent les propriétés du matériau. Bien qu'efficace, cette approche est très coûteuse en calculs à exécuter pour les défauts ponctuels limitant la portée de telles investigations.
"Les calculs fonctionnels de densité fonctionnent bien si vous modélisez une petite unité, mais si vous souhaitez agrandir votre cellule de modélisation, la puissance de calcul requise pour ce faire augmente considérablement, " dit Bharat Medasani, un ancien post-doctorant du Berkeley Lab et auteur principal de l'article npj. "Et parce qu'il est coûteux en calcul de modéliser des défauts dans un seul matériau, faire ce genre de modélisation par force brute pour des dizaines de milliers de matériaux n'est pas faisable."
Pour surmonter ces défis informatiques, Medasani et ses collègues ont développé et formé des algorithmes d'apprentissage automatique pour prédire les défauts ponctuels dans les composés intermétalliques, en se concentrant sur la structure cristalline B2 largement observée. Initialement, ils ont sélectionné un échantillon de 100 de ces composés à partir de la base de données du projet sur les matériaux et ont effectué des calculs fonctionnels de densité sur des superordinateurs au National Energy Research Scientific Computing Center (NERSC), une installation utilisateur du DOE Office of Science au Berkeley Lab, pour identifier leurs défauts.
Parce qu'ils avaient un petit échantillon de données à partir duquel travailler, Medasani et son équipe ont utilisé une approche forestière appelée amplification de gradient pour développer leur méthode d'apprentissage automatique avec une grande précision. Dans cette approche, des modèles d'apprentissage automatique supplémentaires ont été construits successivement et combinés avec des modèles antérieurs pour minimiser la différence entre les prédictions des modèles et les résultats des calculs fonctionnels de densité. Les chercheurs ont répété le processus jusqu'à ce qu'ils atteignent un haut niveau de précision dans leurs prédictions.
"Ce travail est essentiellement une preuve de concept. Il montre que l'on peut faire des calculs fonctionnels de densité pour quelques centaines de matériaux, puis entraînez des algorithmes d'apprentissage automatique pour prédire avec précision les défauts ponctuels pour un groupe de matériaux beaucoup plus important, " dit Medasani, qui est maintenant chercheur postdoctoral au Pacific Northwest National Laboratory.
« L'avantage de ce travail est que nous disposons désormais d'une approche d'apprentissage automatique peu coûteuse en calcul qui peut prédire rapidement et avec précision les défauts ponctuels dans les nouveaux matériaux intermétalliques », déclare Andrew Canning, un scientifique informatique du Berkeley Lab et co-auteur de l'article npj. « Nous n'avons plus à effectuer de calculs de premier principe très coûteux pour identifier les propriétés des défauts de chaque nouveau composé métallique. »
"Cet outil nous permet de prédire les défauts métalliques plus rapidement et de manière robuste, ce qui accélérera à son tour la conception des matériaux, " dit Kristin Persson, un scientifique du Berkeley Lab et directeur du projet Matériaux, une initiative visant à réduire considérablement le temps nécessaire pour inventer de nouveaux matériaux en fournissant un accès Web ouvert aux informations calculées sur les matériaux connus et prévus. Dans le prolongement de ce travail, une boîte à outils Python open source pour la modélisation des défauts ponctuels dans les semi-conducteurs et les isolants (PyCDT) a été développée.