Une équipe de scientifiques informaticiens du laboratoire national d'Oak Ridge du ministère de l'Énergie a généré et publié des ensembles de données d'une ampleur sans précédent qui fournissent les propriétés spectrales visibles dans l'ultraviolet de plus de 10 millions de molécules organiques. Comprendre comment une molécule interagit avec la lumière est essentiel pour découvrir ses propriétés électroniques et optiques, qui à leur tour ont des applications photoactives potentielles dans des produits tels que les cellules solaires ou les systèmes d'imagerie médicale.
À l’aide des ressources informatiques hautes performances du Oak Ridge Leadership Computing Facility, l’équipe ORNL a effectué des calculs de chimie quantique pour générer les vastes ensembles de données. Pour chacune de ces molécules organiques, l’équipe a effectué des calculs de modélisation de matériaux atomistiques avec diverses approximations pour calculer différentes propriétés d’intérêt à l’état excité. Les résultats de l'équipe ont été publiés dans Scientific Data. .
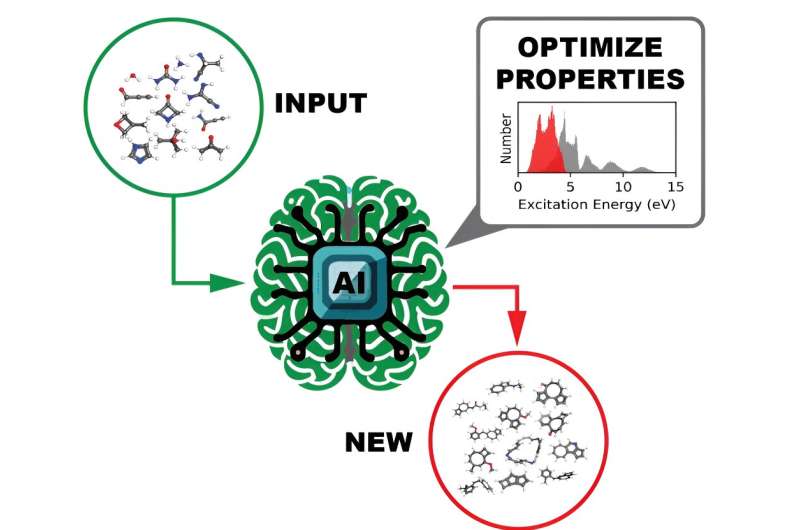
L'utilisation ultime prévue des ensembles de données open source est de former un modèle d'apprentissage profond pour identifier des molécules dotées de propriétés optoélectroniques et de photoréactivité personnalisées, une approche beaucoup plus rapide et plus facile à mettre en œuvre que les méthodes actuelles.
"L'utilisation de modèles DL pour la conception moléculaire est essentielle car l'espace chimique qui doit être exploré pour la recherche de ces molécules est extrêmement vaste", a déclaré l'auteur principal Massimiliano Lupo Pasini, data scientist à la Division des sciences informatiques et de l'ingénierie de l'ORNL.
"Les expériences et les calculs de principes premiers existants, qui sont basés sur les lois physiques qui déterminent la façon dont la matière et l'énergie interagissent au niveau subatomique, sont tout simplement inabordables pour différentes raisons. Les expériences demandent beaucoup de travail, et les calculs de principes premiers peuvent facilement mettre à mal le calcul intensif. Mais les modèles DL fournissent des outils très prometteurs pour surmonter ces obstacles", a déclaré Lupo Pasini.
Le projet a démarré lorsque Stephan Irle, responsable du groupe de chimie computationnelle et de sciences des nanomatériaux de l'ORNL, a identifié les spectres ultraviolet-visible des molécules comme une propriété utile à prédire avec les modèles DL.
Construire un modèle DL suffisamment complexe pour identifier les propriétés moléculaires souhaitables nécessite de l’entraîner avec d’énormes volumes de données explorant toutes les différentes régions de l’espace chimique. Plus il y a de données collectées, plus le modèle DL formé sur celui-ci peut atteindre la robustesse et la généralisabilité nécessaires pour fonctionner efficacement. Cependant, la collecte de volumes aussi importants de données scientifiques pour une DL évolutive peut présenter des problèmes de flux de données, en particulier dans les installations comptant plusieurs utilisateurs comme l'OLCF, une installation utilisateur du Bureau des sciences du DOE située à l'ORNL.
"L'un des défis qui se posent lors de la génération de gros volumes de données est que le nombre de fichiers à gérer augmente considérablement. S'il n'est pas géré correctement, un volume de données aussi important peut compromettre le fonctionnement du système de fichiers parallèle, qui est un élément important du système de fichiers parallèles. Installations HPC de pointe", a déclaré Lupo Pasini.
Pour relever ce défi, Lupo Pasini a collaboré avec l'informaticien Kshitij Mehta de l'ORNL pour développer un logiciel de flux de travail évolutif qui garantit que les fichiers générés par le code de la mécanique quantique sont correctement traités sans stresser le système de fichiers, comme Orion de l'OLCF, qui est un fichier partagé. ressource qui gère l'entrée, la sortie et le stockage des données sur les systèmes de superordinateurs.
À titre de test de validation de principe, l'équipe a généré l'ensemble de données GDB-9-Ex de 96 766 molécules composées de carbone, d'azote, d'oxygène et de fluor, avec au plus neuf atomes non hydrogène. Il a montré que le flux de travail conçu est efficace et que la formation DL prédit avec précision la position et l'intensité des pics les plus pertinents du spectre ultraviolet-visible.
À partir de ce succès initial, l’équipe a augmenté son volume avec l’ensemble de données ORNL_AISD-Ex, qui contient 10 502 917 molécules composées de carbone, d’azote, d’oxygène, de fluor et de soufre, avec au plus 71 atomes non hydrogène. Pilsun Yoo, associé de recherche postdoctoral dans le groupe d'Irle, a développé des outils pour analyser les ensembles de données résultants.
Le spectre ultraviolet-visible, qui décrit les modes d'excitation d'une molécule, a été calculé pour chacune des plus de 10 millions de molécules. Ces informations révèlent quelle fréquence lumineuse est nécessaire pour cibler une molécule et briser certaines liaisons du composé chimique.
Une autre propriété intéressante calculée pour chaque molécule était l’écart HOMO-LUMO – l’écart énergétique entre l’orbitale moléculaire occupée la plus élevée et l’orbitale moléculaire inoccupée la plus basse – qui mesure de manière fiable la stabilité de la molécule. Grâce à ces informations, un modèle DL pourrait efficacement passer au crible les données pour identifier des molécules prometteuses pour différentes utilisations prospectives.
En fait, Lupo Pasini et son équipe de l'ORNL, dont le spécialiste en informatique en apprentissage automatique Pei Zhang et le chercheur en données HPC Jong Youl Choi, développent précisément un tel modèle DL :HydraGNN.
"L'architecture HydraGNN prend en compte la structure atomique, la convertit en un graphique, puis essaie de prédire comme résultat ce que produirait le code des premiers principes. Il s'agit d'un modèle de substitution pour les calculs coûteux des premiers principes", a déclaré Lupo Pasini.
Les résultats de la formation d'HydraGNN sur les ensembles de données et ses découvertes moléculaires seront détaillés dans un prochain article.
Plus d'informations : Massimiliano Lupo Pasini et al, Deux ensembles de données à l'état excité pour les spectres chimiques quantiques UV-visible de molécules organiques, Données scientifiques (2023). DOI :10.1038/s41597-023-02408-4
Informations sur le journal : Données scientifiques
Fourni par le Laboratoire national d'Oak Ridge