Les chercheurs ont développé une plateforme qui combine des expériences automatisées avec l'IA pour prédire comment les produits chimiques réagiront les uns avec les autres, ce qui pourrait accélérer le processus de conception de nouveaux médicaments.
Prédire la façon dont les molécules réagiront est vital pour la découverte et la fabrication de nouveaux produits pharmaceutiques, mais historiquement, cela a été un processus d'essais et d'erreurs, et les réactions échouent souvent. Pour prédire la réaction des molécules, les chimistes simulent généralement les électrons et les atomes dans des modèles simplifiés, un processus coûteux en calcul et souvent inexact.
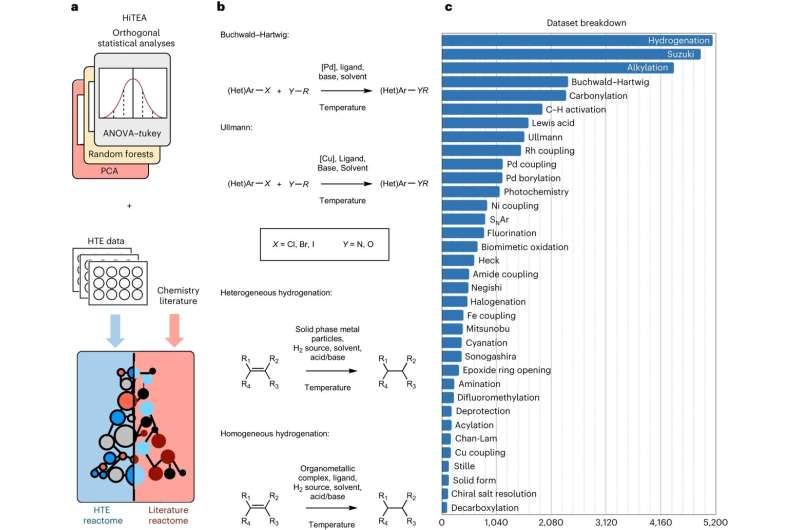
Aujourd’hui, des chercheurs de l’Université de Cambridge ont développé une approche basée sur les données, inspirée de la génomique, où des expériences automatisées sont combinées à l’apprentissage automatique pour comprendre la réactivité chimique, accélérant ainsi considérablement le processus. Ils ont appelé leur approche, qui a été validée sur un ensemble de données de plus de 39 000 réactions pharmaceutiquement pertinentes, le « reactome » chimique.
Leurs résultats, rapportés dans la revue Nature Chemistry , sont le fruit d'une collaboration entre Cambridge et Pfizer.
"Le réactome pourrait changer notre façon de penser la chimie organique", a déclaré le Dr Emma King-Smith du laboratoire Cavendish de Cambridge, premier auteur de l'article. "Une compréhension plus approfondie de la chimie pourrait nous permettre de fabriquer des produits pharmaceutiques et bien d'autres produits utiles beaucoup plus rapidement. Mais plus fondamentalement, la compréhension que nous espérons générer sera bénéfique à tous ceux qui travaillent avec des molécules."
L'approche du réactome sélectionne les corrélations pertinentes entre les réactifs, les réactifs et les performances de la réaction à partir des données, et souligne les lacunes dans les données elles-mêmes. Les données sont générées à partir d'expériences automatisées très rapides ou à haut débit.
"La chimie à haut débit a changé la donne, mais nous pensions qu'il existait un moyen de découvrir une compréhension plus profonde des réactions chimiques que ce qui peut être observé à partir des premiers résultats d'une expérience à haut débit", a déclaré King-Smith.
"Notre approche révèle les relations cachées entre les composants de la réaction et les résultats", a déclaré le Dr Alpha Lee, qui a dirigé la recherche. "L'ensemble de données sur lequel nous avons formé le modèle est énorme :il contribuera à faire passer le processus de découverte chimique de l'essai et de l'erreur à l'ère du Big Data."
Dans un article connexe, publié dans Nature Communications , l'équipe a développé une approche d'apprentissage automatique qui permet aux chimistes d'introduire des transformations précises dans des régions moléculaires prédéfinies, permettant ainsi une conception plus rapide de médicaments.
Cette approche permet aux chimistes de modifier des molécules complexes, comme un changement de conception de dernière minute, sans avoir à les créer à partir de zéro. La fabrication d’une molécule en laboratoire est généralement un processus en plusieurs étapes, comme la construction d’une maison. Si les chimistes veulent modifier le noyau d'une molécule, la méthode conventionnelle consiste à reconstruire la molécule, comme si on démolirait la maison et qu'on la reconstruirait à partir de zéro. Cependant, les variations fondamentales sont importantes pour la conception des médicaments.
Une classe de réactions connues sous le nom de réactions de fonctionnalisation à un stade avancé tente d'introduire directement des transformations chimiques dans le noyau, évitant ainsi de devoir repartir de zéro. Cependant, il est difficile de rendre sélective et contrôlée la fonctionnalisation à un stade avancé :de nombreuses régions des molécules peuvent généralement réagir, et il est difficile de prédire le résultat.
"Les fonctionnalisations à un stade avancé peuvent produire des résultats imprévisibles et les méthodes de modélisation actuelles, y compris notre propre intuition d'expert, ne sont pas parfaites", a déclaré King-Smith. "Un modèle plus prédictif nous donnerait la possibilité d'effectuer un meilleur dépistage."
Les chercheurs ont développé un modèle d’apprentissage automatique qui prédit où une molécule réagirait et comment le site de réaction varierait en fonction de différentes conditions de réaction. Cela permet aux chimistes de trouver des moyens de modifier avec précision le noyau d'une molécule.
"Nous avons pré-entraîné le modèle sur un grand nombre de données spectroscopiques, enseignant efficacement la chimie générale du modèle, avant de l'affiner pour prédire ces transformations complexes", a déclaré King-Smith. Cette approche a permis à l’équipe de surmonter la limitation du faible niveau de données :il existe relativement peu de réactions de fonctionnalisation à un stade avancé rapportées dans la littérature scientifique. L'équipe a validé expérimentalement le modèle sur un ensemble diversifié de molécules de type médicament et a pu prédire avec précision les sites de réactivité dans différentes conditions.
"L'application de l'apprentissage automatique à la chimie est souvent limitée par le problème de la petite quantité de données par rapport à l'immensité de l'espace chimique", a déclaré Lee. "Notre approche, qui consiste à concevoir des modèles qui apprennent à partir de grands ensembles de données similaires mais différents du problème que nous essayons de résoudre, résout ce défi fondamental lié aux faibles données et pourrait débloquer des avancées au-delà de la fonctionnalisation à un stade avancé."
Plus d'informations : Emma King-Smith et al, Sonder le « réactome » chimique avec des données d'expérimentation à haut débit, Nature Chemistry (2024). DOI :10.1038/s41557-023-01393-w
Fonctionnalisation prédictive de Minisci à un stade avancé avec apprentissage par transfert, Nature Communications (2024). DOI :10.1038/s41467-023-42145-1. www.nature.com/articles/s41467-023-42145-1
Informations sur le journal : Communications naturelles , Chimie naturelle
Fourni par l'Université de Cambridge