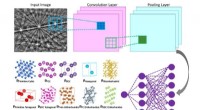
Une équipe de recherche de l'Institut de chimie organique et de biochimie de l'Académie tchèque des sciences/IOCB Prague a développé une nouvelle méthode informatique capable de décrire avec précision comment les protéines interagissent avec les molécules de médicaments potentiels et peuvent le faire en quelques dizaines de minutes seulement. Cette nouvelle fonction de notation de la mécanique quantique peut ainsi accélérer considérablement la recherche de nouveaux médicaments. La recherche a été publiée dans la revue Nature Communications .
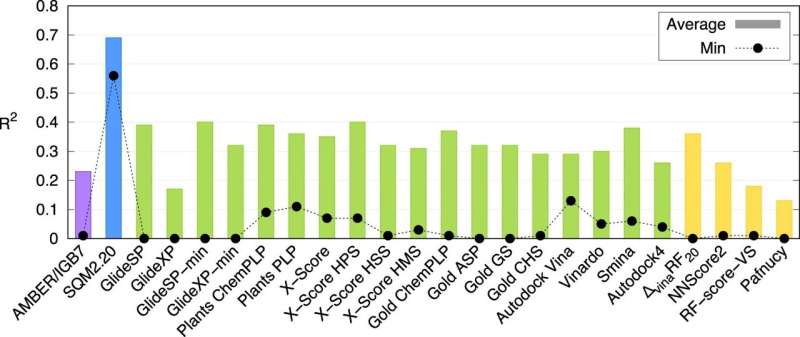
L’étude démontre qu’il s’agit de la première méthode universellement applicable de ce type. Les experts en informatique de l'IOCB Prague l'ont testé sur 10 protéines de différents niveaux de complexité structurelle, chacune se liant à une grande variété de petites molécules (généralement appelées ligands). Ils ont ensuite comparé leurs résultats non seulement avec ceux d'autres méthodes correspondantes, mais aussi avec les résultats d'expériences en laboratoire, et les deux comparaisons se sont révélées très favorables.
"Bien sûr, nous ne sommes pas les seuls à travailler sur ce sujet. Il existe plusieurs méthodes de ce type. Cependant, leur rapidité est généralement compensée par une faible précision alors que des calculs plus précis peuvent prendre plusieurs jours. Nos méthodes sont uniques dans le sens où elles peuvent traiter des informations sur de grands systèmes moléculaires en quelques dizaines de minutes tout en conservant les avantages de calculs de mécanique quantique beaucoup plus exigeants", explique Jan Řezáč, auteur correspondant de l'article du groupe Interactions non covalentes dirigé par le professeur Pavel Hobza.
Les experts de ce groupe étudient depuis longtemps les interactions intermoléculaires. Dans ces recherches, ils se concentrent principalement sur les biomolécules, et les résultats de leurs travaux portent directement sur la conception assistée par ordinateur de médicaments. La raison en est que lorsque les scientifiques travaillent sur un nouveau médicament, ils recherchent souvent des molécules qui se lient fortement à une protéine particulière.
Cependant, les identifier revient à trouver des aiguilles dans une botte de foin, car il faut tester un grand nombre de molécules pour distinguer celles qui sont prometteuses. Cela ralentit considérablement la découverte de substances médicinales et la rend plus coûteuse. En prédisant la force de liaison protéine-ligand, et en sélectionnant ainsi les molécules qui satisfont le mieux à un ensemble défini de critères, les chimistes computationnels épargnent le travail des expérimentateurs, ce qui, à son tour, accélère considérablement la découverte de médicaments.
Plus d'informations : Adam Pecina et al, SQM2.20 :La fonction de notation semi-empirique de mécanique quantique produit des prédictions d'affinité de liaison protéine-ligand de qualité DFT en quelques minutes, Nature Communications (2024). DOI :10.1038/s41467-024-45431-8
Informations sur le journal : Communications naturelles
Fourni par l'Institut de chimie organique et de biochimie du CAS