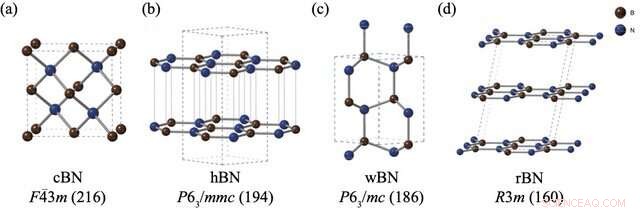
Les structures et les groupes spatiaux de (a) le nitrure de bore de mélange de zinc (cBN), (b) le nitrure de bore hexagonal (hBN), (c) le nitrure de bore wurtzite (wBN) et (d) le nitrure de bore rhomboédrique (rBN). Les atomes de bore et d'azote sont représentés respectivement en marron et en bleu. Crédit :Kousuke Nakano de JAIST.
Le nitrure de bore (BN) est un matériau polyvalent avec des applications dans une variété de domaines techniques et scientifiques. Cela est dû en grande partie à une propriété intéressante du BN appelée "polymorphisme", caractérisée par la capacité à se cristalliser en plus d'un type de structure. Cela se produit généralement en réponse à des changements de température, de pression ou des deux. De plus, les différentes structures, appelées "polymorphes", diffèrent remarquablement dans leurs propriétés physiques malgré la même formule chimique. En conséquence, les polymorphes jouent un rôle important dans la conception des matériaux, et une connaissance de la manière de favoriser sélectivement la formation du polymorphe souhaité est cruciale à cet égard.
Cependant, les polymorphes BN posent un problème particulier. Malgré la réalisation de plusieurs expériences pour évaluer les stabilités relatives des polymorphes BN, un consensus n'a pas émergé sur ce sujet. Alors que les méthodes de calcul sont souvent l'approche de choix pour ces problèmes, les polymorphes BN ont posé de sérieux défis aux techniques de calcul standard en raison des faibles "interactions de van der Waals (vdW)" entre leurs couches, qui ne sont pas prises en compte dans ces calculs. De plus, les quatre polymorphes BN stables, à savoir rhomboédrique (rBN), hexagonal (hBN), wurtzite (wBN) et zinc-blende (cBN), se manifestent dans une plage d'énergie étroite, ce qui permet de capturer de petites différences d'énergie avec les interactions vdW encore plus difficile.
Une équipe de recherche internationale dirigée par le professeur adjoint Kousuke Nakano du Japan Advanced Institute of Science and Technology (JAIST) a maintenant fourni des preuves pour régler le débat. Dans leur étude, ils ont abordé le problème avec un cadre de calcul de premier principe à la pointe de la technologie, à savoir des simulations Monte Carlo de diffusion à nœud fixe (FNDMC). FNDMC représente une étape dans la méthode populaire de simulation quantique de Monte Carlo, dans laquelle une "fonction d'onde" quantique à plusieurs corps paramétrée est d'abord optimisée pour atteindre l'état fondamental, puis fournie au FNDMC.
De plus, l'équipe a également calculé l'énergie de Gibbs (le travail utile pouvant être obtenu à partir d'un système à pression et température constantes) des polymorphes BN pour différentes températures et pressions en utilisant la théorie de la fonctionnelle de la densité (DFT) et des calculs de phonons. Cet article a été mis en ligne le 24 mars 2022 et publié dans The Journal of Physical Chemistry C .
Selon les résultats du FNDMC, hBN était la structure la plus stable, suivie de rBN, cBN et wBN. Ces résultats étaient cohérents à la fois à 0 K et à 300 K (température ambiante). Cependant, les estimations DFT ont donné des résultats contradictoires pour deux approximations différentes. Le Dr Nakano explique ces résultats contradictoires :"Nos résultats révèlent que l'estimation des stabilités relatives est fortement influencée par la fonctionnelle corrélationnelle d'échange, ou l'approximation utilisée dans le calcul DFT. Par conséquent, une conclusion quantitative ne peut pas être tirée en utilisant les résultats DFT, et une approche plus précise, telle que FNDMC, est nécessaire."
Notamment, les résultats de FNDMC étaient en accord avec ceux générés par d'autres méthodes de calcul raffinées, telles que le "cluster couplé", suggérant que FNDMC est un outil efficace pour traiter les polymorphes, en particulier ceux régis par les forces vdW. L'équipe a également montré qu'elle peut fournir d'autres informations importantes, telles que des énergies de référence fiables, lorsque les données expérimentales ne sont pas disponibles.
Le Dr Nakano est enthousiasmé par les perspectives d'avenir de la méthode dans le domaine de la science des matériaux. "Notre étude démontre la capacité du FNDMC à détecter de minuscules changements d'énergie impliquant des forces vdW, ce qui stimulera l'utilisation de cette méthode pour d'autres matériaux de van der Waals", dit-il. "De plus, les simulations moléculaires basées sur cette méthode précise et fiable pourraient permettre la conception de matériaux, permettant le développement de médicaments et de catalyseurs." Augmenter la précision des calculs de force atomique avec la transformation des coordonnées de distorsion spatiale