Résumé graphique. Crédit :Journal of Chemical Information and Modeling (2022). DOI :10.1021/acs.jcim.2c00389
La symétrie est une caractéristique répandue de la nature à toutes les échelles. Par exemple, nos yeux nus peuvent facilement identifier les symétries dans la forme corporelle d'innombrables organismes. La symétrie est également très importante dans les domaines de la physique et de la chimie, en particulier dans le domaine microscopique des atomes et des molécules. Les cristaux, qui sont des matériaux hautement ordonnés, peuvent même avoir plusieurs types de symétrie en même temps, comme la symétrie de rotation, la symétrie d'inversion et la symétrie de translation.
Dernièrement, parallèlement aux progrès rapides de l'informatique, les chercheurs ont développé des méthodes de calcul qui cherchent à prédire les propriétés physiques des cristaux en fonction de leur structure électronique. En pratique, cependant, les cristaux purs et parfaitement symétriques sont rarement utilisés. En effet, les propriétés d'un cristal peuvent être ajustées à volonté en les alliant avec d'autres matériaux ou en remplaçant au hasard certains atomes par d'autres éléments, c'est-à-dire par dopage.
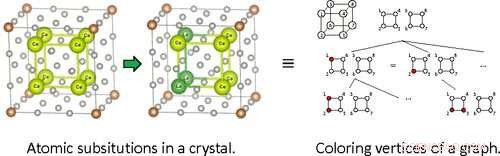
En conséquence, les scientifiques des matériaux recherchent des approches informatiques efficaces pour analyser ces alliages et cristaux substitués, également connus sous le nom de solutions solides. La «méthode des supercellules» est une de ces approches et est largement utilisée pour modéliser des structures cristallines avec des substitutions aléatoires de différents atomes. La symétrie des cristaux, cependant, est en fait un problème lors de l'utilisation de cette technique. Dans les cristaux, il peut y avoir de nombreux modèles de substitution qui sont physiquement équivalents à d'autres substitutions si nous les traduisons ou les faisons simplement pivoter. Trouver ces modèles de substitution symétriques n'est pas très significatif, et donc leur calcul lors de l'utilisation de la méthode des supercellules est une perte de temps.
Dans une étude récente, une équipe de chercheurs dirigée par le professeur adjoint Kousuke Nakano du Japan Advanced Institute of Science and Technology (JAIST) a trouvé une solution à ce problème. Ils ont développé un logiciel open source appelé "Suite pour la génération à haut débit de modèles avec des substitutions atomiques implémentées par Python" ou SHRY qui peut, en termes de symétrie, générer des modèles de substitution distincts dans des solutions solides et des alliages. Ce travail, qui a été publié dans le Journal of Chemical Information and Modeling , a été co-écrit par le doctorant Genki I. Prayogo, le Dr Andrea Tirelli, le professeur Ryo Maezono et le professeur associé Kenta Hongo.
L'équipe a abordé le problème sous l'angle de la théorie des groupes. Il s'avère que la recherche de modèles de substitution atomique dans les cristaux est analogue au problème de la recherche de modèles de coloration sur les sommets des graphes sous certaines restrictions. Cela permet de reformuler le problème original de trouver des substitutions atomiques non symétriques dans les cristaux en explorant des arbres de recherche décrivant la coloration des sommets dans les graphes.
Cependant, la manière dont l'arbre de recherche est exploré est cruciale. Une approche simple et naïve dans laquelle toutes les branches possibles sont recherchées et directement comparées est impossible; le temps et les calculs nécessaires augmentent de manière incontrôlable pour les grands systèmes. Cela se produit parce que décider d'explorer ou non une branche plus loin nécessite des informations sur toutes les autres branches en plus de celle qui est explorée, ce qui est techniquement appelé "informations non locales".
Pour éviter ce problème, les chercheurs ont implémenté dans SHRY une technique appelée augmentation canonique. "Cette méthode peut décider si une branche d'arbre doit être explorée plus profondément ou non en se basant uniquement sur des informations locales", explique le Dr Nakano, "Plus important encore, les théorèmes de la théorie des groupes garantissent que seuls des modèles de substitution distincts seront extraits, sans sur- ou sous-explorant la structure arborescente en termes de symétrie." L'équipe a vérifié que leur algorithme était sans erreur en le testant minutieusement avec des données provenant d'une base de données de structures cristallines.
Il convient de noter que SHRY a été écrit en Python 3, l'un des langages de programmation multiplateformes les plus populaires, et téléchargé sur GitHub, une plateforme en ligne de partage de projets de premier plan. « SHRY peut être utilisé en tant que programme autonome ou importé dans un autre programme Python en tant que module », souligne le Dr Nakano, « Notre logiciel utilise également le format de fichier d'informations cristallographiques (CIF) largement pris en charge pour l'entrée et la sortie du ensembles de structures cristallines substituées." L'équipe prévoit de continuer à améliorer le code de SHRY en fonction des commentaires des autres utilisateurs, en augmentant sa vitesse et ses capacités.
Dans l'ensemble, le logiciel développé dans cette étude pourrait aider les scientifiques à identifier les substitutions atomiques potentielles dans les solides, ce qui est la stratégie la plus couramment utilisée pour ajuster les propriétés des matériaux pour des applications pratiques. SHRY contribuera à accélérer la recherche et à développer des cristaux de substitution dotés de fonctionnalités sans précédent et de caractéristiques supérieures. Polaritons de cisaillement hyperpoliques dans des cristaux de faible symétrie