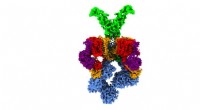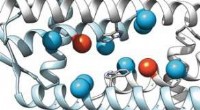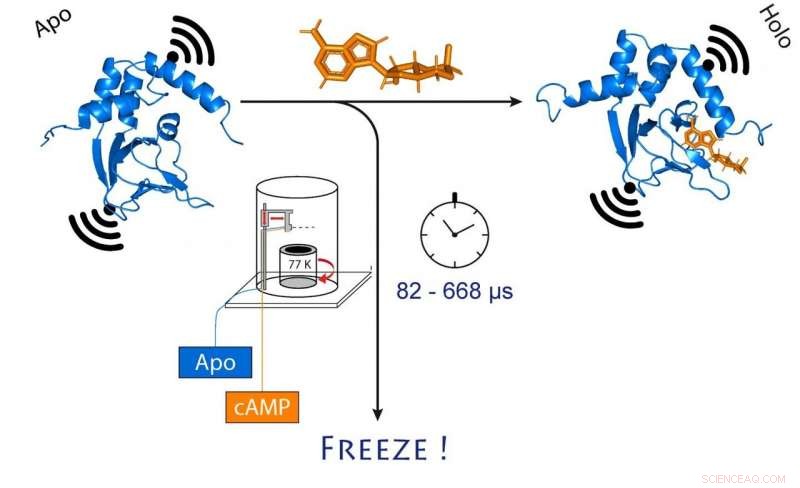
lorsque l'AMPc (orange) se lie. Cela modifie également la distance entre les deux aimants moléculaires attachés au canal (noir). Crédit :(c) Tobias Hett / Universität Bonn und Forschungszentrum césar
Des chercheurs de l'Université de Bonn et du Research Center Caesar ont réussi à congeler des protéines ultra-rapides après une période de temps définie avec précision. Ils ont pu suivre les changements structurels à l'échelle de la microseconde et avec une précision inférieure au nanomètre. En raison de sa haute résolution spatiale et temporelle, la méthode permet de suivre les changements structurels rapides des enzymes et des acides nucléiques. Les résultats sont publiés dans le Journal de l'American Chemical Society.
Si vous voulez savoir à quoi ressemble la structure spatiale d'une biomolécule, vous disposez d'un formidable arsenal d'outils. Les plus populaires sont la microscopie électronique et la diffraction des rayons X, qui peut révéler même les plus petits détails d'une protéine. Cependant, une limitation importante de ces méthodes est qu'elles fournissent généralement des images statiques, qui sont souvent insuffisants pour comprendre les processus biomoléculaires en termes mécanistiques précis. Par conséquent, un objectif à long terme de nombreux groupes de recherche dans le monde a été de suivre les mouvements au sein d'une macromolécule telle qu'une protéine au fil du temps pendant qu'elle effectue son travail, comme dans un film. Les groupes de recherche dirigés par le professeur Dr. Olav Schiemann de l'Institut de chimie physique et théorique de l'Université de Bonn et le professeur Dr. Benjamin Kaupp du Centre de recherche César de la Société Max Planck ont fait un pas de plus vers cet objectif. but.
Ils ont choisi un canal ionique pour leur enquête. Il s'agit d'une protéine qui forme de minuscules pores dans la membrane cellulaire qui sont perméables aux particules chargées appelées ions. "Ce canal est normalement fermé, " explique Schiemann. " Il ne s'ouvre que lorsqu'un messager cellulaire, appelé AMPc, s'y lie. Nous voulions savoir comment fonctionnait exactement ce processus."
Mini aimants pour mesurer les distances
Faire cela, les chercheurs ont d'abord mélangé la protéine du canal et l'AMPc, puis ont rapidement congelé la solution. A l'état congelé, la structure de la protéine peut maintenant être analysée. Pour que leur méthode fonctionne, ils avaient attaché des électro-aimants moléculaires en deux points du canal. La distance entre ces aimants peut être déterminée avec une précision de quelques Angström (dix milliardièmes de millimètre) en utilisant une méthode sophistiquée appelée PELDOR, qui fonctionne comme une règle moléculaire. Dans les années récentes, la méthode a été considérablement affinée et améliorée dans le groupe de Schiemann.
"Toutefois, cela ne nous donne qu'une image statique de la liaison de l'AMPc au canal ionique, " dit Schiemann. " Nous avons donc répété le processus de congélation à différents moments après avoir mélangé les deux molécules. Cela a permis de reconstruire les mouvements de la protéine après la liaison à l'AMPc, comme dans un film, qui est également constitué d'une séquence d'images."
Au centre de cette procédure se trouve une méthode sophistiquée qui permet de mélanger et de congeler les échantillons très rapidement à un moment précis. La technique, appelé "microseconde gel hypertrempe" (en abrégé MHQ), a été développé à l'origine à l'Université de Delft, mais tomba plus tard en désuétude. Il a été redécouvert et affiné de manière décisive par le groupe de Kaupp.
"Dans l'appareil MHQ, la molécule d'AMPc et le canal ionique sont mélangés à vitesse ultrarapide, " explique Kaupp. " Ensuite, le mélange est projeté en un mince filet sur un cylindre métallique très froid à -190 °C, qui tourne 7, 000 fois par minute. Il était particulièrement difficile de transférer les échantillons congelés pour la mesure PELDOR de la plaque métallique dans des tubes de verre minces, et de les garder congelés en attendant. Nous avons dû concevoir et construire des outils spéciaux pour cela."
Congélation en 82 millionièmes de seconde
L'ensemble du processus de mélange et de congélation ne prend que 82 microsecondes (une microseconde équivaut à un millionième de seconde). « Cela nous permet de visualiser des changements très rapides dans la structure spatiale des protéines, " explique Tobias Hett, l'un des deux doctorants qui ont contribué de manière significative à la réussite. L'avantage de la méthode est sa combinaison de haute résolution spatiale et temporelle. "Cela représente une avancée majeure dans l'étude des processus dynamiques dans les biomolécules, ", souligne Kaupp.
Les chercheurs prévoient maintenant d'utiliser leur méthode pour examiner de plus près d'autres biomolécules. Ils espèrent acquérir de nouvelles connaissances, par exemple dans le fonctionnement des enzymes et des acides nucléiques. L'importance de telles informations est mieux illustrée par la récente vague mondiale de recherche structurelle sur le coronavirus-2 du SRAS :la soi-disant protéine de pointe du virus subit également un changement structurel lorsque les cellules humaines sont infectées. Clarifier ce mécanisme fournira des informations précieuses sur la façon de cibler le mécanisme d'infection avec de nouveaux médicaments.
La préparation des échantillons, l'exécution expérimentale, et l'analyse des données est très complexe. Les résultats de l'étude reflètent donc également une coopération scientifique réussie avec des chercheurs dirigés par le professeur Dr. Helmut Grubmüller de l'Institut Max Planck de chimie biophysique à Göttingen et le professeur Dr. Heinz-Jürgen Steinhoff de l'Université d'Osnabrück.