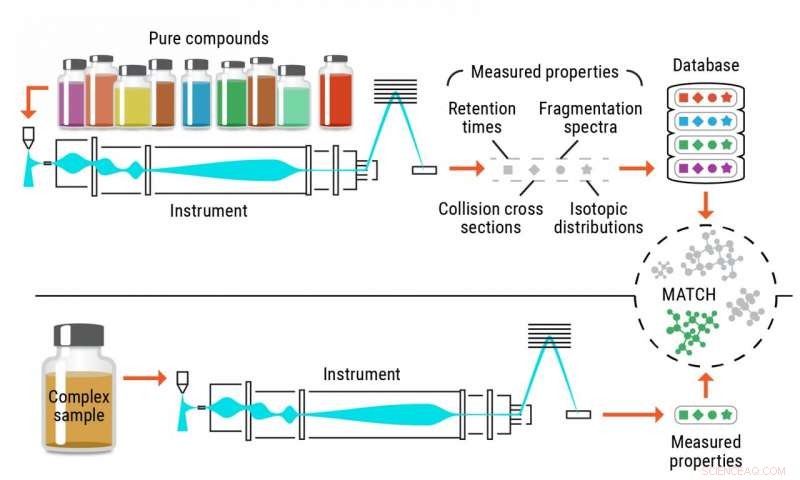
Illustration du processus classique d'identification des métabolites. Crédit :Laboratoire national du Nord-Ouest du Pacifique
Identification précise des métabolites, et d'autres petits produits chimiques, dans les échantillons biologiques et environnementaux a toujours été insuffisant lors de l'utilisation de méthodes traditionnelles. Les tactiques conventionnelles reposent sur des composés de référence purs, appelés normes, reconnaître les mêmes molécules dans des échantillons complexes. Ces approches sont limitées par la disponibilité des produits chimiques purs qui sont utilisés comme normes.
« Nous voulions vraiment contourner le paradigme actuel de la conduite d'une expérience de métabolomique et de la façon dont les molécules sont identifiées en toute confiance, " dit Tom Metz, scientifique biomédical au Pacific Northwest National Laboratory (PNNL) et directeur du Pacific Northwest Advanced Compound Identification Core.
Un problème avec la méthode actuelle est qu'il n'y a qu'un nombre limité de composés purs que les chercheurs peuvent acheter auprès de fournisseurs; la plupart des fournisseurs ont accès à environ 3, 000–4, 000 composés.
"Si vous considérez ce qui devrait se produire dans la nature, tu regardes> 1030 composés ou plus qui pourraient être possibles, " dit Metz. " Alors, lorsque vous comparez les quelques milliers de produits chimiques standard auxquels vous avez accès avec le grand nombre de composés potentiels, tu n'es même pas proche."
Approche d'identification sans normes
Pour résoudre ce problème, Metz et son équipe du PNNL ont conceptualisé une approche - la métabolomique sans normes - avec laquelle ils calculent ou prédisent des informations sur plusieurs propriétés pour les molécules d'intérêt afin de générer des bibliothèques de référence complètes, puis de faire correspondre les données expérimentales contenant les mêmes propriétés à ces bibliothèques, permettant l'identification des composés.
Grâce à cette nouvelle approche, les chercheurs envoient des structures chimiques via des programmes d'apprentissage automatique ou de chimie quantique pour prédire avec précision les propriétés expérimentales des métabolites.
"Si nous sommes suffisamment précis sur ces prédictions, nous n'aurions théoriquement plus jamais besoin d'analyser un composé pur, " a déclaré Metz. " Cette collection d'outils va changer le paradigme actuel de la métabolomique, et dans un avenir proche, il y aura de très bonnes applications pour montrer à la communauté des chercheurs les avantages de cette nouvelle approche. »
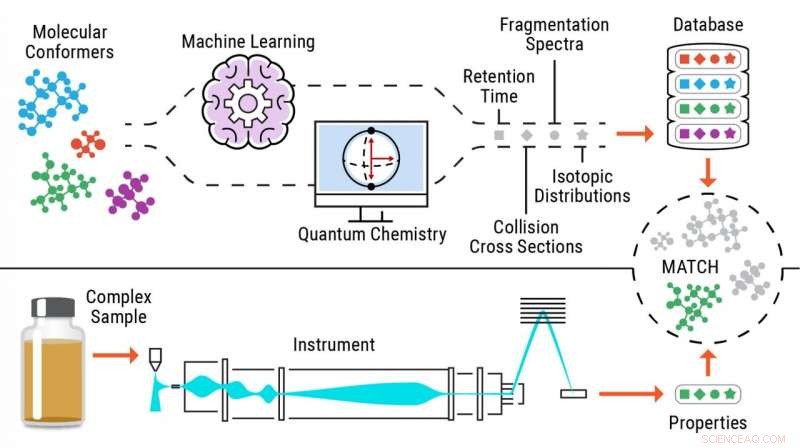
Illustration du processus d'identification sans normes des métabolites. Crédit :Laboratoire national du Nord-Ouest du Pacifique
En n'ayant pas à se fier aux données d'analyses d'étalons purs pour identifier les petites molécules, l'approche sans normes permet d'identifier jusqu'à 90 % de produits chimiques en plus dans les échantillons et rend ces outils de calcul très utiles dans plusieurs domaines d'application, y compris la découverte de nouveaux médicaments, médecine légale, et la recherche environnementale et biomédicale.
"Par exemple, dans la conception d'un nouveau médicament, un utilisateur pourrait dire, « J'ai un certain nombre de propriétés avec ces certains médicaments, mais ils se trouvent être toxiques. Pouvons-nous prédire un composé qui aurait des propriétés similaires mais qui pourrait ne pas être toxique ? », a déclaré Metz. « Si les bonnes données d'entraînement pouvaient être fournies au programme DarkChem, DarkChem pourrait alors effectuer cette prédiction."
Suite de programmes personnalisable
La nouvelle approche de l'identification métabolomique sans normes utilise quatre outils clés pour générer des dans des bibliothèques de référence de métabolites dérivés de silico, et pour extraire et faire correspondre les données expérimentales pour obtenir des identifications de composés :
Les outils ont été conçus pour fonctionner ensemble, mais ils peuvent également être utilisés séparément. Les chercheurs peuvent personnaliser les différentes applications en fonction des besoins ou des domaines de recherche d'un client, créant une approche complètement modulaire.
Faire avancer un domaine de recherche
À l'heure actuelle, dans la communauté métabolomique, tous les chercheurs identifient le même ensemble de molécules dans chaque échantillon. La raison en est qu'ils ont tous les mêmes composés purs qu'ils ont achetés pour construire leurs bibliothèques de référence.
"Notre vision est qu'en utilisant l'approche sans normes, vous ne serez jamais limité par l'étendue des petites molécules pouvant être identifiées dans un échantillon, ", a déclaré Metz. "C'est vraiment un changeur de jeu pour la métabolomique. Et c'est très excitant de voir ce que l'année prochaine nous réserve."