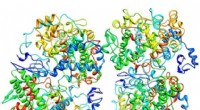
Les résultats d'une analyse de réseau de neurones artificiels (ANN) peuvent ne pas être fiables pour des molécules trop différentes de celles sur lesquelles l'ANN a été formé. Les nuages noirs montrés ici couvrent des complexes de métaux de transition dans l'ensemble de données dont les représentations numériques sont trop éloignées de celles des complexes d'entraînement pour être considérées comme fiables. Crédit :Massachusetts Institute of Technology
Dans les années récentes, L'apprentissage automatique s'est avéré être un outil précieux pour identifier de nouveaux matériaux avec des propriétés optimisées pour des applications spécifiques. Travailler avec de grands, des ensembles de données bien définis, les ordinateurs apprennent à effectuer une tâche analytique pour générer une réponse correcte, puis à utiliser la même technique sur un ensemble de données inconnu.
Bien que cette approche ait guidé le développement de nouveaux matériaux précieux, il s'agit principalement de composés organiques, note Heather Kulik Ph.D. '09, professeur adjoint de génie chimique. Kulik se concentre plutôt sur les composés inorganiques, en particulier, ceux à base de métaux de transition, une famille d'éléments (dont le fer et le cuivre) qui ont des propriétés uniques et utiles. Dans ces composés, connus sous le nom de complexes de métaux de transition, l'atome de métal se trouve au centre avec des bras liés chimiquement, ou des ligands, en carbone, hydrogène, azote, ou des atomes d'oxygène rayonnant vers l'extérieur.
Les complexes de métaux de transition jouent déjà un rôle important dans des domaines allant du stockage d'énergie à la catalyse pour la fabrication de produits chimiques fins, par exemple, pour les produits pharmaceutiques. Mais Kulik pense que l'apprentissage automatique pourrait encore étendre leur utilisation. En effet, son groupe a travaillé non seulement pour appliquer l'apprentissage automatique à l'inorganique - une entreprise nouvelle et stimulante - mais aussi pour utiliser la technique pour explorer de nouveaux territoires. "Nous voulions comprendre jusqu'où nous pouvions pousser nos modèles pour faire des découvertes, pour faire des prédictions sur des composés qui n'avaient jamais été vus auparavant, " dit Kulik.
Capteurs et ordinateurs
Depuis quatre ans, Kulik et Jon Paul Janet, un étudiant diplômé en génie chimique, se sont concentrés sur les complexes de métaux de transition avec « spin », une propriété de mécanique quantique des électrons. D'habitude, les électrons se produisent par paires, un avec spin up et l'autre avec spin down, donc ils s'annulent et il n'y a pas de spin net. Mais dans un métal de transition, les électrons peuvent être non appariés, et le spin net résultant est la propriété qui fait des complexes inorganiques d'intérêt, dit Kulik. « Adapter à quel point les électrons ne sont pas appariés nous donne un bouton unique pour adapter les propriétés. »
Un complexe donné a un état de spin préféré. Mais ajoutez un peu d'énergie, disons, de la lumière ou de la chaleur - et il peut basculer dans l'autre état. Dans le processus, il peut présenter des changements dans les propriétés macroscopiques telles que la taille ou la couleur. Lorsque l'énergie nécessaire pour provoquer le retournement, appelée énergie de séparation de spin, est proche de zéro, le complexe est un bon candidat pour une utilisation comme capteur, ou peut-être en tant que composant fondamental d'un ordinateur quantique.
Les chimistes connaissent de nombreuses combinaisons métal-ligand avec des énergies de séparation de spin proches de zéro, ce qui en fait des complexes potentiels "spin-crossover" (SCO) pour de telles applications pratiques. Mais l'ensemble des possibilités est vaste. L'énergie de séparation de spin d'un complexe de métal de transition est déterminée par les ligands qui sont combinés avec un métal donné, et il existe une infinité de ligands parmi lesquels choisir. Le défi consiste à trouver de nouvelles combinaisons avec la propriété souhaitée pour devenir des SCO, sans recourir à des millions de tests et d'erreurs en laboratoire.
Traduire des molécules en nombres
La méthode standard pour analyser la structure électronique des molécules est d'utiliser une méthode de modélisation informatique appelée théorie fonctionnelle de la densité, ou DFT. Les résultats d'un calcul DFT sont assez précis, en particulier pour les systèmes organiques, mais effectuer un calcul pour un seul composé peut prendre des heures, voire des jours. En revanche, un outil d'apprentissage automatique appelé réseau de neurones artificiels (ANN) peut être formé pour effectuer la même analyse, puis le faire en quelques secondes seulement. Par conséquent, Les ANN sont beaucoup plus pratiques pour rechercher des SCO possibles dans l'immense espace des complexes réalisables.

Ce graphique représente un échantillon de complexe de métal de transition. Un complexe de métal de transition se compose d'un atome central de métal de transition (orange) entouré d'un ensemble de molécules organiques liées chimiquement dans des structures connues sous le nom de ligands. Crédit :Massachusetts Institute of Technology
Parce qu'un ANN nécessite une entrée numérique pour fonctionner, le premier défi des chercheurs était de trouver un moyen de représenter un complexe de métaux de transition donné sous la forme d'une série de nombres, chacun décrivant une propriété sélectionnée. Il existe des règles pour définir les représentations des molécules organiques, où la structure physique d'une molécule en dit long sur ses propriétés et son comportement. Mais lorsque les chercheurs ont suivi ces règles pour les complexes de métaux de transition, ça n'a pas marché. "La liaison métal-organique est très délicate à réaliser, " dit Kulik. " Il y a des propriétés uniques de la liaison qui sont plus variables. Les électrons peuvent choisir de nombreuses autres manières pour former une liaison. » Les chercheurs ont donc dû élaborer de nouvelles règles pour définir une représentation qui serait prédictive en chimie inorganique.
En utilisant l'apprentissage automatique, ils ont exploré diverses façons de représenter un complexe de métaux de transition pour analyser l'énergie de séparation de spin. Les résultats étaient meilleurs lorsque la représentation mettait le plus l'accent sur les propriétés du centre métallique et la connexion métal-ligand et moins sur les propriétés des ligands plus éloignés. De façon intéressante, leurs études ont montré que les représentations qui accordaient une importance plus égale dans l'ensemble fonctionnaient mieux lorsque l'objectif était de prédire d'autres propriétés, telles que la longueur de la liaison ligand-métal ou la tendance à accepter des électrons.
Tester l'ANN
Pour tester leur approche, Kulik et Janet, assistés de Lydia Chan, un stagiaire d'été de Troy High School à Fullerton, Californie—définit un ensemble de complexes de métaux de transition basés sur quatre métaux de transition—chrome, manganèse, fer à repasser, et cobalt—dans deux états d'oxydation avec 16 ligands (chaque molécule peut en avoir jusqu'à deux). En combinant ces blocs de construction, ils ont créé un "espace de recherche" de 5, 600 complexes, dont certains familiers et bien étudiés, et certains d'entre eux totalement inconnus.
Dans des travaux antérieurs, les chercheurs avaient formé un ANN sur des milliers de composés bien connus dans la chimie des métaux de transition. Pour tester la capacité de l'ANN formé à explorer un nouvel espace chimique pour trouver des composés avec les propriétés ciblées, ils ont essayé de l'appliquer au pool de 5, 600 ensembles, 113 dont il avait vu dans l'étude précédente.
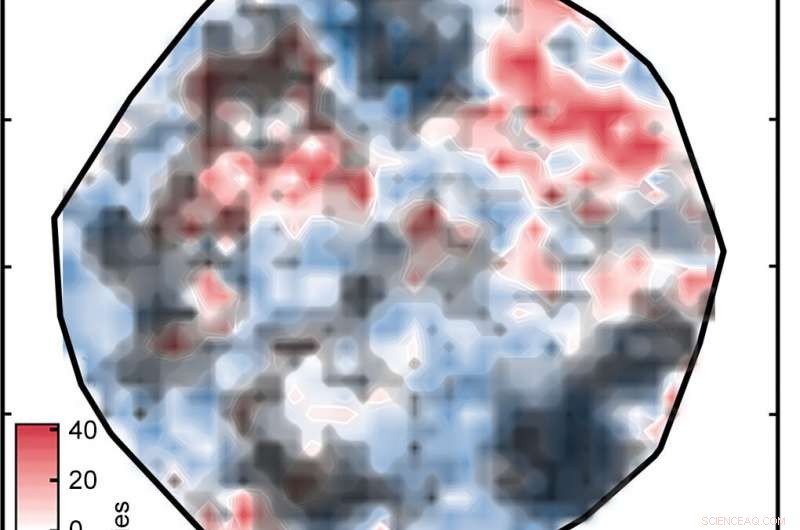
Le résultat a été le tracé intitulé "Figure 1" dans le diaporama ci-dessus, qui trie les complexes sur une surface telle que déterminée par l'ANN. Les régions blanches indiquent des complexes avec des énergies de séparation de spin à moins de 5 kilocalories par mole de zéro, ce qui signifie qu'ils sont potentiellement de bons candidats SCO. Les régions rouges et bleues représentent des complexes avec des énergies de séparation de spin trop grandes pour être utiles. Les losanges verts qui apparaissent dans l'encart montrent des complexes qui ont des centres de fer et des ligands similaires - en d'autres termes, composés apparentés dont les énergies de croisement de spin devraient être similaires. Leur apparition dans une même région de la parcelle témoigne de la bonne correspondance entre la représentation des chercheurs et les propriétés clés du complexe.
Mais il y a un hic :toutes les prédictions de fractionnement de spin ne sont pas exactes. Si un complexe est très différent de ceux sur lesquels le réseau a été formé, l'analyse ANN peut ne pas être fiable - un problème standard lors de l'application de modèles d'apprentissage automatique à la découverte en science des matériaux ou en chimie, note Kulik. En utilisant une approche qui semblait fructueuse dans leurs travaux précédents, les chercheurs ont comparé les représentations numériques des complexes d'entraînement et de test et ont exclu tous les complexes de test où la différence était trop importante.
Se concentrer sur les meilleures options
Effectuer l'analyse ANN de tous les 5, 600 complexes n'ont pris qu'une heure. Mais dans le monde réel, le nombre de complexes à explorer pourrait être des milliers de fois plus grand et tout candidat prometteur nécessiterait un calcul DFT complet. Les chercheurs avaient donc besoin d'une méthode d'évaluation d'un grand ensemble de données pour identifier les candidats inacceptables avant même l'analyse ANN. À cette fin, ils ont développé un algorithme génétique – une approche inspirée de la sélection naturelle – pour noter les complexes individuels et éliminer ceux jugés inaptes.
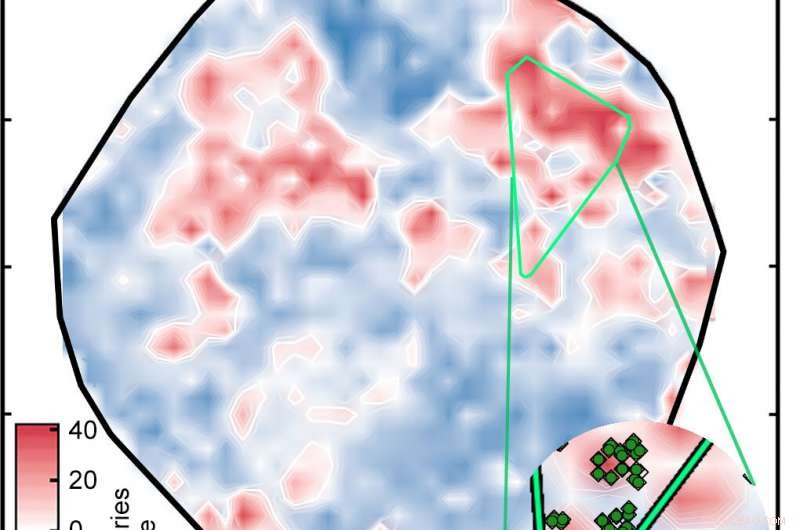
Un réseau de neurones artificiels préalablement entraînés sur des composés bien connus analysés 5, 600 complexes de métaux de transition pour identifier les complexes potentiels de spin-crossover. Le résultat fut ce complot, dans lequel les complexes sont colorés en fonction de leur énergie de séparation de spin en kilocalories par mole (kcal/mol). Chez les candidats prometteurs, cette énergie est à moins de 5 kcal/mol de zéro. Les diamants vert vif dans l'encart sont des complexes apparentés. Crédit :Massachusetts Institute of Technology
Pour présélectionner un ensemble de données, l'algorithme génétique sélectionne d'abord au hasard 20 échantillons parmi l'ensemble complet des complexes. Il attribue ensuite un score de « fitness » à chaque échantillon sur la base de trois mesures. D'abord, son énergie de croisement de spin est-elle suffisamment faible pour qu'il soit un bon SCO ? Découvrir, le réseau de neurones évalue chacun des 20 complexes. Seconde, le complexe est-il trop éloigné des données d'entraînement ? Si c'est le cas, l'énergie de croisement de spin de l'ANN peut être inexacte. Et enfin, le complexe est-il trop proche des données d'entraînement ? Si c'est le cas, les chercheurs ont déjà effectué un calcul DFT sur une molécule similaire, le candidat n'a donc pas d'intérêt dans la recherche de nouvelles options.
Sur la base de son évaluation en trois parties des 20 premiers candidats, l'algorithme génétique rejette les options inaptes et enregistre le plus apte pour le prochain tour. Pour assurer la diversité des composés sauvegardés, l'algorithme demande à certains d'entre eux de muter un peu. Un complexe peut se voir attribuer un nouveau, ligand choisi au hasard, ou deux complexes prometteurs peuvent échanger des ligands. Après tout, si un complexe a l'air bien, alors quelque chose de très similaire pourrait être encore mieux - et le but ici est de trouver de nouveaux candidats. L'algorithme génétique ajoute alors de nouvelles, complexes choisis au hasard pour remplir le deuxième groupe de 20 et effectue sa prochaine analyse. En répétant ce processus 21 fois au total, il produit 21 générations d'options. Il parcourt ainsi l'espace de recherche, permettre aux candidats les plus aptes de survivre et de se reproduire, et les inaptes à mourir.
Effectuer l'analyse de 21 générations sur les 5 complets, Ensemble de 600 données complexes requis un peu plus de cinq minutes sur un ordinateur de bureau standard, et il a donné 372 pistes avec une bonne combinaison de diversité élevée et de confiance acceptable. Les chercheurs ont ensuite utilisé DFT pour examiner 56 complexes choisis au hasard parmi ces pistes, et les résultats ont confirmé que les deux tiers d'entre eux pourraient être de bons SCO.
Bien qu'un taux de réussite de deux tiers puisse sembler peu élevé, les chercheurs font deux remarques. D'abord, leur définition de ce qui pourrait faire un bon SCO était très restrictive :pour qu'un complexe survive, son énergie de séparation de spin devait être extrêmement faible. Et deuxieme, étant donné un espace de 5, 600 complexes et rien d'autre, combien d'analyses DFT seraient nécessaires pour trouver 37 pistes ? Comme le note Janet, "Peu importe combien nous en avons évalué avec le réseau de neurones, car c'est tellement bon marché. Ce sont les calculs DFT qui prennent du temps."
Le meilleur de tous, l'utilisation de leur approche a permis aux chercheurs de trouver des candidats SCO non conventionnels auxquels on n'aurait pas pensé sur la base de ce qui a été étudié dans le passé. "Il y a des règles que les gens ont - des heuristiques dans leur tête - pour savoir comment ils construiraient un complexe de spin-crossover, ", explique Kulik. "Nous avons montré que vous pouvez trouver des combinaisons inattendues de métaux et de ligands qui ne sont normalement pas étudiées mais qui peuvent être prometteuses en tant que candidats au spin-crossover."
Partager les nouveaux outils
Pour soutenir la recherche mondiale de nouveaux matériaux, les chercheurs ont incorporé l'algorithme génétique et l'ANN dans « molSimplify, " le groupe est en ligne, boîte à outils logicielle open source que tout le monde peut télécharger et utiliser pour construire et simuler des complexes de métaux de transition. Pour aider les utilisateurs potentiels, le site propose des didacticiels qui montrent comment utiliser les fonctionnalités clés des codes logiciels open source. Le développement de molSimplify a commencé avec un financement de la MIT Energy Initiative en 2014, et tous les étudiants du groupe de Kulik y ont contribué depuis.
Les chercheurs continuent d'améliorer leur réseau neuronal pour étudier les SCO potentiels et publier des versions mises à jour de molSimplify. Pendant ce temps, d'autres dans le laboratoire de Kulik développent des outils qui peuvent identifier des composés prometteurs pour d'autres applications. Par exemple, un domaine d'intérêt important est la conception de catalyseurs. L'étudiant diplômé en chimie Aditya Nandy se concentre sur la recherche d'un meilleur catalyseur pour convertir le méthane en un carburant liquide plus facile à manipuler comme le méthanol, un problème particulièrement difficile. "Maintenant, nous avons une molécule extérieure qui arrive, et notre complexe - le catalyseur - doit agir sur cette molécule pour effectuer une transformation chimique qui se déroule en toute une série d'étapes, " dit Nandy. " L'apprentissage automatique sera très utile pour déterminer les paramètres de conception importants pour un complexe de métaux de transition qui rendra chaque étape de ce processus énergétiquement favorable. "
Cette histoire est republiée avec l'aimable autorisation de MIT News (web.mit.edu/newsoffice/), un site populaire qui couvre l'actualité de la recherche du MIT, innovation et enseignement.