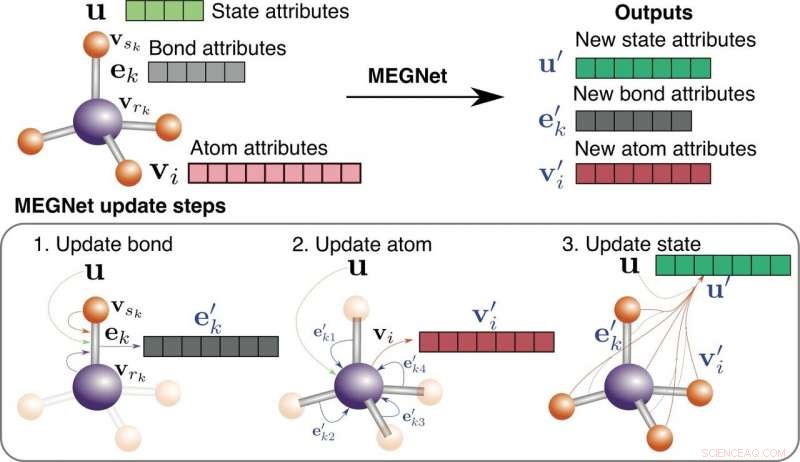
Illustration schématique des modèles MEGNet. Crédit :Chi Chen/Laboratoire virtuel des matériaux
Des nano-ingénieurs de l'Université de Californie à San Diego ont développé de nouveaux modèles d'apprentissage en profondeur qui peuvent prédire avec précision les propriétés des molécules et des cristaux. En permettant des prédictions de propriétés quasi instantanées, ces modèles d'apprentissage en profondeur offrent aux chercheurs les moyens d'analyser rapidement l'univers presque infini des composés pour découvrir des matériaux potentiellement transformateurs pour diverses applications technologiques, telles que les batteries Li-ion à haute densité d'énergie, LED blanc chaud, et une meilleure photovoltaïque.
Pour construire leurs modèles, une équipe dirigée par le professeur de nano-ingénierie Shyue Ping Ong à la Jacobs School of Engineering de l'UC San Diego a utilisé un nouveau cadre d'apprentissage en profondeur appelé réseaux de graphes, développé par Google DeepMind, le cerveau derrière AlphaGo et AlphaZero. Les réseaux de graphes ont le potentiel d'étendre les capacités de la technologie d'IA existante pour effectuer des tâches d'apprentissage et de raisonnement complexes avec une expérience et des connaissances limitées, ce pour quoi les humains sont bons.
Pour les scientifiques des matériaux comme Ong, les réseaux de graphes offrent un moyen naturel de représenter les relations de liaison entre les atomes d'une molécule ou d'un cristal et permettent aux ordinateurs d'apprendre comment ces relations sont liées à leurs propriétés chimiques et physiques.
Les nouveaux modèles basés sur des réseaux de graphes, que l'équipe d'Ong a surnommé les modèles MatErials Graph Network (MEGNet), a surpassé l'état de l'art en prédisant 11 propriétés sur 13 pour les 133, 000 molécules dans le jeu de données QM9. L'équipe a également formé les modèles MEGNet sur environ 60, 000 cristaux dans le Projet Matériaux. Les modèles ont surpassé les modèles d'apprentissage automatique antérieurs pour prédire les énergies de formation, bandes interdites et modules d'élasticité des cristaux.
L'équipe a également démontré deux approches pour surmonter les limitations des données en science des matériaux et en chimie. D'abord, l'équipe a montré que les réseaux de graphes peuvent être utilisés pour unifier plusieurs modèles d'énergie libre, résultant en une augmentation multiple des données d'entraînement. Seconde, ils ont montré que leurs modèles MEGNet peuvent apprendre efficacement les relations entre les éléments du tableau périodique. Ces informations apprises par machine à partir d'un modèle de propriété formé sur un grand ensemble de données peuvent ensuite être transférées pour améliorer la formation et la précision des modèles de propriété avec de plus petites quantités de données. Ce concept est connu dans l'apprentissage automatique sous le nom d'apprentissage par transfert.