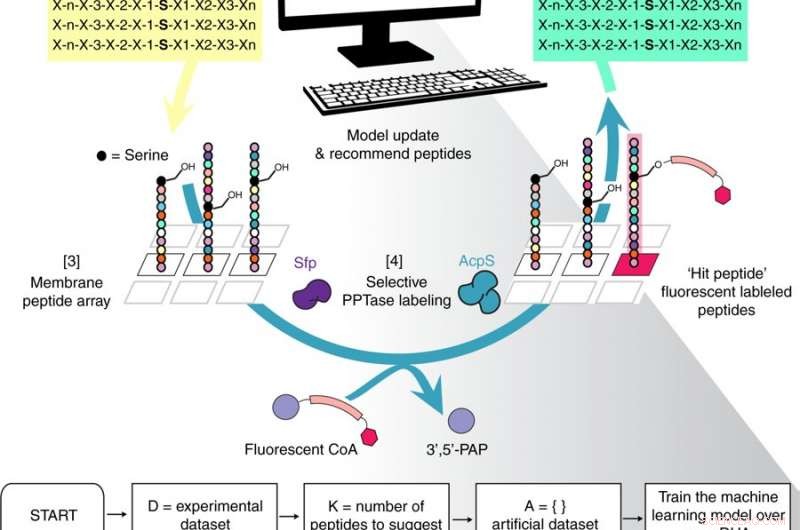
Présentation du flux de travail de la méthode itérative Peptide Optimization with Optimal Learning (POOL). Crédit: Communication Nature (2018). DOI :10.1038/s41467-018-07717-6
Les scientifiques et les ingénieurs s'intéressent depuis longtemps à la synthèse de peptides, des chaînes d'acides aminés responsables de nombreuses fonctions au sein des cellules, à la fois pour imiter la nature et pour effectuer de nouvelles activités. Un peptide conçu, par exemple, pourrait être un médicament fonctionnel agissant dans certaines zones du corps sans se dégrader, une tâche difficile pour de nombreux peptides.
Mais les méthodes de découverte et de synthèse de peptides sont coûteuses et chronophages, impliquant souvent des mois ou des années de conjectures et d'échecs.
Chercheurs de l'Université Northwestern, en s'associant avec des collaborateurs de l'Université Cornell et de l'Université de Californie, San Diego, ont développé une nouvelle façon de trouver des séquences peptidiques optimales :en utilisant un algorithme d'apprentissage automatique en tant que collaborateur.
L'algorithme analyse les données expérimentales et propose des suggestions sur la prochaine meilleure séquence à essayer, créant un processus de sélection aller-retour qui réduit considérablement le temps nécessaire pour trouver le peptide optimal.
Les résultats, qui pourrait fournir un nouveau cadre pour les expériences en science des matériaux et en chimie, ont été publiés dans Communication Nature le 7 décembre.
"Nous considérons cela comme la prochaine vague dans la façon dont nous concevons des molécules et des matériaux, " a déclaré le professeur du Nord-Ouest Nathan Gianneschi, un auteur correspondant sur le papier. "Nous pouvons combiner ce que nous savons de l'intuition avec la puissance d'un algorithme et trouver la solution avec moins d'expériences."
Gianneschi est professeur Jacob et Rosaline Cohn au département de chimie du Weinberg College of Arts and Sciences de Northwestern et aux départements de science et d'ingénierie des matériaux et de génie biomédical de Northwestern Engineering.
Pour créer la méthode, Gianneschi, qui est également directeur associé de l'Institut international de nanotechnologie de Northwestern, fait équipe avec Peter Frazier, un professeur agrégé à Cornell qui travaille dans la recherche opérationnelle et l'apprentissage automatique, et Michel Burkart, biologiste chimique et expert en enzymologie à l'UC San Diego, pour trouver une meilleure façon de fabriquer des peptides qui pourraient générer des biomatériaux, en particulier des nanostructures et des microstructures qui pourraient modifier les protéines de certaines manières. La première étape consistait à trouver les bons peptides qui agiraient comme substrats enzymatiques pour ces structures.
Les peptides sont construits à partir de chaînes d'acides aminés pouvant aller jusqu'à 20 acides aminés, avec 20 possibilités différentes pour chaque acide. Puisque la séquence détermine la fonction peptidique, déterminer les séquences optimales nécessite des expériences coûteuses souvent menées avec des conjectures.
Les expérimentateurs, Gianneschi et Burkart, a travaillé avec Frazier pendant plusieurs années pour développer un système qui combinait des données expérimentales avec un algorithme d'apprentissage automatique pour trouver les meilleures stratégies pour créer de nouveaux matériaux.
Après que Frazier ait conçu l'algorithme et que les deux aient travaillé ensemble pour l'entraîner, les expérimentateurs ont développé une puce de 100 peptides, mené des expériences pour déterminer celles qui fonctionnaient comme elles étaient censées, puis introduit ces informations dans l'algorithme. L'algorithme a ensuite recommandé ce qu'il faut changer pour le prochain cycle de développement de peptides, et a également recommandé des stratégies qui, selon lui, échoueraient.
« Maintenant, nous commencions à avoir de la sélectivité, " a déclaré Gianneschi. En complétant ce processus plusieurs fois, ils ont pu se concentrer sur les peptides optimaux.
"Au lieu de deviner et de regarder des millions de peptides, nous avons pu observer des centaines de peptides et converger très rapidement sur des séquences au comportement totalement nouveau, " a-t-il dit. Comparé à des mutations aléatoires ou à des conjectures, la méthode de l'algorithme a été statistiquement beaucoup plus réussie.
Bien que ce travail se soit concentré sur les substrats, ce processus pourrait être utilisé pour découvrir des peptides pour tout type de but, comme l'administration de médicaments, et peut-être même être utilisé pour découvrir des séquences d'ADN, également. Parce que toute sorte de séquence optimale pourrait être découverte, les chercheurs ne sont pas non plus limités aux séquences d'acides aminés trouvées dans le code génétique.
La prochaine étape consistera à automatiser l'ensemble du processus. Gianneschi s'intéresse également à l'utilisation de la méthode pour trouver des surfaces optimales pour les polymères, en particulier les polymères utilisés dans les implants médicaux. Trouver les bonnes surfaces qui se lieront aux tissus ou aux muscles pourrait aider à prévenir le tissu cicatriciel ou le rejet de l'implant.
"Vous pourriez essentiellement découvrir des séquences qui font des choses spécifiques, qui est vraiment au cœur de ce que font les peptides et les acides nucléiques dans la nature, ", a-t-il déclaré. "Cela pourrait révolutionner la façon dont nous fabriquons des peptides."