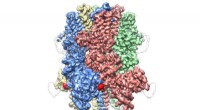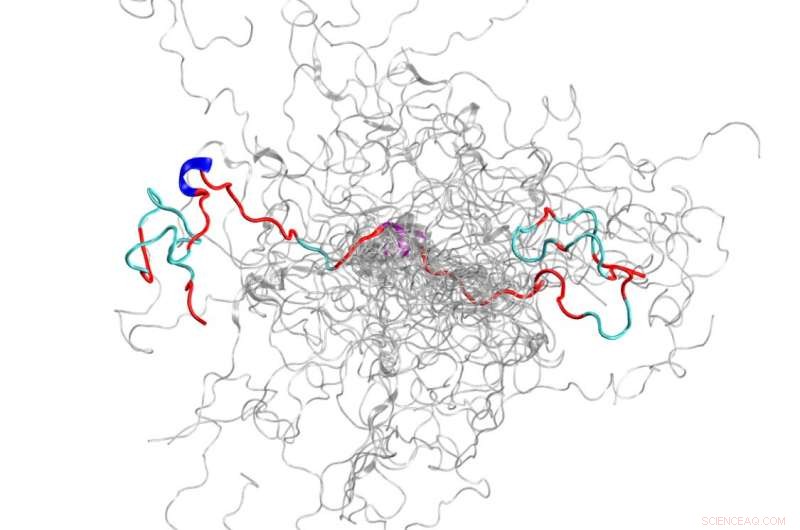
L'ensemble configurationnel (une collection de structures 3D) d'une protéine intrinsèquement désordonnée, l'extrémité N-terminale de la c-Src kinase, qui est une protéine de signalisation majeure chez l'homme. Crédit :Laboratoire national d'Oak Ridge, Département américain de l'énergie
À l'aide du supercalculateur Titan et de la source de neutrons de spallation du laboratoire national d'Oak Ridge du ministère de l'Énergie, les scientifiques ont créé le modèle 3D le plus précis à ce jour d'une protéine intrinsèquement désordonnée, révélant l'ensemble de ses structures au niveau atomique.
Comme son nom l'indique, un IDP n'adopte pas un ordre, structure statique comme les autres protéines; au lieu, il est flexible et peut adopter plusieurs structures 3-D. Ce manque d'une structure unique est nécessaire pour la fonction biologique de l'IDP, mais le rend techniquement difficile à étudier. Les IDP peuvent être une protéine entière ou un domaine d'une protéine autrement structurée, et ils constituent une grande partie de l'homme, microbe, et les protéines végétales.
Loukas Petridis, chercheur au Centre de biophysique moléculaire de l'ORNL, a dirigé une équipe de chercheurs vers une nouvelle façon de créer des modèles physiques précis de ces biosystèmes flexibles, ce qui peut conduire à une meilleure compréhension de leurs fonctions biologiques. Au cours des trois dernières années, l'équipe a combiné des expériences de diffusion de neutrons avec des simulations améliorées de dynamique moléculaire (MD) d'échantillonnage si exigeantes en termes de calcul qu'elles nécessitaient la puissance de traitement de Titan, le Cray XK7 à 27 pétaflops récemment mis hors service au Oak Ridge Leadership Computing Facility, une installation d'utilisateurs du DOE Office of Science à l'ORNL.
« Étudier ces personnes déplacées est assez difficile, tant du point de vue de l'expérimentation que de la modélisation, " dit Utsab Shrestha, l'auteur principal de l'article de l'équipe, récemment publié dans le Actes de l'Académie nationale des sciences . "Nous n'y avons pas seulement pensé à partir de l'expérience ou de la simulation, nous avons prévu de mettre en synergie ces deux approches – de les combiner de manière à obtenir des informations plus précises sur les personnes déplacées. Spécifiquement, les simulations nous ont aidés à générer un ensemble précis d'IDP à résolution atomique, ce qui est difficile à déterminer à partir d'expériences seules.
Typiquement, les chercheurs mènent des expériences telles que la diffusion de neutrons aux petits angles, diffusion des rayons X aux petits angles, ou la résonance magnétique nucléaire pour sonder des systèmes biologiques flexibles. Cependant, ces méthodes ne fournissent pas une image détaillée au niveau atomique des structures 3-D d'un IDP, connu comme son ensemble configurationnel. Par ailleurs, ils ne peuvent produire que des données moyennes d'ensemble, plutôt que les configurations spécifiques de la structure des protéines sous-jacentes. Les scientifiques ont également effectué des simulations informatiques d'IDP et les ont comparées à de telles expériences, dans l'espoir d'obtenir les mêmes résultats afin de vérifier l'exactitude de leurs modèles.
"Mais ils finissent par ne pas être d'accord avec les expériences, " dit Petridis. " Et à cause de la divergence entre les simulations et les expériences, ils doivent repondérer les simulations - ils doivent ajuster les résultats de simulation pour les faire correspondre aux expériences, ce qui est frustrant. C'était l'état de l'art jusqu'à notre travail."
Les simulations MD par ordinateur menées par Shrestha ont utilisé des méthodes d'échantillonnage améliorées qui ont réussi à faire correspondre non seulement les expériences de diffusion de neutrons - menées par Viswanathan Gurumoorthy et ses collègues du SNS, une installation utilisateur du DOE Office of Science à l'ORNL, mais également des données RMN précédemment publiées. Ces simulations MD utilisent la physique pour déterminer comment les protéines se déplacent. La clé du succès de l'équipe a été d'exécuter de nombreuses simulations MD en parallèle sur Titan, permettant aux simulations de communiquer entre elles et d'échanger des informations.
"C'est très important car cela permet à la simulation d'échantillonner un plus grand espace de configuration, explorer davantage les structures tridimensionnelles de manière plus efficace, " a déclaré Petridis. "C'est pourquoi ce MD à échantillonnage amélioré peut produire des résultats que la simulation MD normale ne peut pas. Nous devrions exécuter une simulation MD normale pendant des années pour obtenir les mêmes résultats. »
L'IDP que l'équipe a choisi d'étudier est le domaine N-terminal de la kinase c-Src, qui est une protéine de signalisation majeure chez l'homme. Des mutations dans cette protéine complexe ont été corrélées avec le cancer, ce qui en fait également une cible médicamenteuse importante. Lors de la cartographie de ce domaine auparavant trouble, les scientifiques ont pu découvrir de nouvelles informations sur ses structures 3-D que les méthodes précédentes n'avaient pas montrées. Par exemple, bien qu'il soit en grande partie désordonné, cette protéine forme des structures ordonnées transitoires, comme les hélices.
"La combinaison d'expériences de diffusion de neutrons et de simulation est très puissante, " a déclaré Petridis. " La validation des simulations par rapport aux expériences de diffusion de neutrons est essentielle pour avoir confiance dans les résultats de la simulation. Les simulations validées peuvent alors fournir des informations détaillées qui ne sont pas directement obtenues par des expériences. »
Le modèle informatique détaillé de l'ensemble de structures 3D de l'IDP ouvre la porte à plus d'expérimentation. Par exemple, les scientifiques pourraient simuler l'effet de la phosphorylation (l'ajout d'un groupe phosphate à la protéine qui peut réguler la fonction de la protéine) pour voir quels changements structurels se produisent dans la kinase c-Src qui pourraient influencer sa fonction. Le rôle des mutations pourrait également être examiné :si un chercheur modifie un acide aminé dans la chaîne, en quoi cela affecte-t-il la structure ou l'ensemble des structures ?
« Il y a beaucoup de questions sans réponse pour la kinase c-Src en particulier qui pourraient être résolues en termes d'interactions avec d'autres partenaires - l'effet de la phosphorylation, l'effet des mutations, " a déclaré Petridis.
Au-delà des utilisations scientifiques potentielles du modèle lui-même, Petridis voit des opportunités d'appliquer l'utilisation du calcul haute performance pour exécuter un échantillonnage amélioré MD pour étudier les structures de nombreux autres IDP importants, ce qui pourrait donner un aperçu de leur fonction. Et plus largement, l'équipe souhaite développer des technologies de simulation capables de reproduire des profils de diffusion de neutrons aux petits angles de systèmes biologiques encore plus complexes.
"Nous ne voulons pas étudier uniquement les protéines désordonnées - nous voulons avoir des systèmes beaucoup plus grands qui contiennent des domaines ordonnés et désordonnés qui peuvent interagir avec les membranes ou l'ADN, " dit Petridis. " La diffusion des neutrons est, à mon avis, la meilleure technique expérimentale pour sonder ces systèmes à plusieurs composants, par exemple, une protéine qui interagit avec une membrane ou une protéine qui interagit avec l'ADN. Mais, toujours, la diffusion des neutrons a besoin de simulations précises pour mieux interpréter les données."