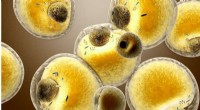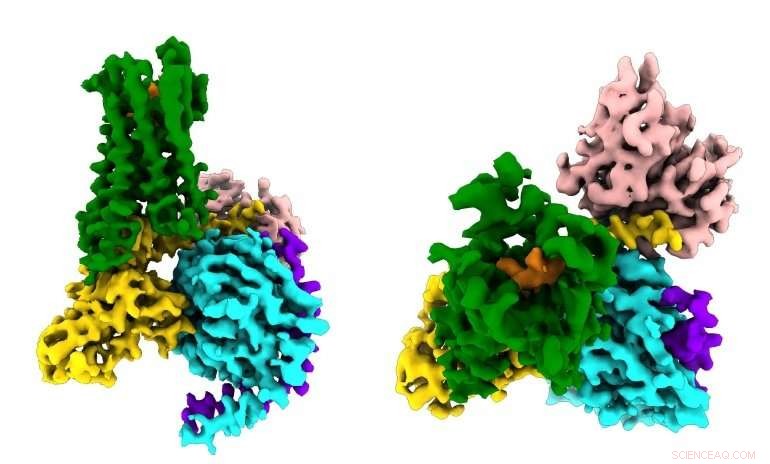
Les chercheurs voient pour la première fois comment un médicament opioïde synthétique (orange) se lie aux récepteurs µ-opioïdes (vert) dans le cerveau, et activer des molécules de signalisation au sein des neurones (G⍺s en or, Gβ en cyan, Gγ en violet) qui conduisent à la suppression de la douleur et à la dépendance. Crédit :Antoine Koehl (Laboratoire Manglik)
Les médicaments opioïdes comme la morphine et le fentanyl sont un pilier de la médecine moderne de la douleur. Mais ils provoquent aussi la constipation, sont très addictifs, et peut entraîner une insuffisance respiratoire mortelle s'il est pris à une dose trop élevée. Les scientifiques cherchent depuis longtemps à développer de nouveaux médicaments opioïdes qui peuvent chasser la douleur sans ces effets secondaires dangereux, mais des lacunes dans notre compréhension de la manière exacte dont les opioïdes exercent leurs divers effets au niveau biologique ont jusqu'à présent tenu ce rêve à distance.
Les analgésiques opioïdes agissent en se liant à une protéine réceptrice présente sur les cellules nerveuses appelée récepteur µ-opioïde, qui a évolué pour répondre aux analgésiques naturels du corps (comme les endorphines produites par l'exercice) en atténuant la douleur et en créant un sentiment d'euphorie. Les opioïdes, de l'opium à la morphine en passant par l'héroïne, détournent ce système de signalisation en se liant à la même molécule réceptrice. Mais les détails sur la façon dont l'activation de ces récepteurs déclenche les effets positifs et négatifs des médicaments sont restés flous.
Maintenant, dans une étude publiée le 13 juin, 2018 en La nature , des scientifiques de l'UC San Francisco et de l'Université de Stanford ont utilisé la cryo-microscopie électronique à ultra-haute résolution (cryoEM) pour capturer le portrait le plus détaillé jamais réalisé d'un médicament opioïde déclenchant la cascade de signalisation biochimique qui lui donne son pouvoir, à la fois pour le meilleur et pour le pire. .
"Nous avons essentiellement capturé cet événement de signalisation dans l'acte, " a déclaré le co-auteur principal de l'étude Aashish Manglik, MARYLAND, Doctorat., un professeur adjoint de chimie pharmaceutique à l'École de pharmacie de l'UCSF qui a mené la nouvelle étude en tant qu'étudiant diplômé et membre distingué à Stanford. "Ces nouvelles images au niveau atomique nous permettront, espérons-le, de concevoir rationnellement des composés qui ciblent différents aspects de la signalisation opioïde dans le cerveau, dans l'espoir d'identifier de nouveaux, des analgésiques plus sûrs."
Le récepteur µ-opioïde fait partie d'une grande famille de centaines de protéines de signalisation appelées récepteurs couplés aux protéines G (RCPG) qui sont impliquées dans tout, de la vision et de l'audition à la réponse du système immunitaire aux agents pathogènes invasifs, et sont les cibles de plus de 30 pour cent des médicaments modernes. La plupart des GPCR partagent les mêmes mécanismes de base :Lorsque la bonne molécule de signalisation (par exemple, un opioïde) se lie à un GPCR à l'extérieur de la cellule, la protéine stimule une réaction en chaîne de signaux biochimiques au sein de la cellule en activant une molécule messagère appelée protéine G (d'où le nom GPCR).
Des expériences qui ont révélé comment un type différent de GPCR se lie à la protéine G "stimulatrice" ont conduit à un prix Nobel pour Brian Kobilka de Stanford, MARYLAND, l'un des auteurs principaux de la nouvelle étude, mais les chercheurs savent depuis des décennies que les GPCR peuvent également se lier à une douzaine d'autres molécules de signalisation au sein de la cellule. Par exemple, Les récepteurs µ-opioïdes n'activent généralement que les protéines G dites « inhibitrices », qui ont l'effet inverse de la cascade stimulatrice de la protéine G. Cependant, les scientifiques ne sont pas sûrs de ce qui cause l'affinité de certains GPCR pour des protéines partenaires particulières dans la cellule, ou exactement quelles sont les conséquences.
Les chercheurs espèrent qu'en comprenant ces différentes voies de signalisation GPCR, ils peuvent être capables de développer des médicaments aux effets très spécifiques, comme supprimer la douleur sans provoquer de dépendance. Mais jusqu'à maintenant, les chercheurs savaient peu comment un GPCR donné interagit sélectivement avec seulement un sous-ensemble de partenaires de signalisation au sein de la cellule.
La nouvelle étude, publié le 13 juin 2018 en La nature , capturé pour la première fois comment le récepteur µ-opioïde se lie à son partenaire inhibiteur de la protéine G. Entre autres constatations, l'étude a montré que la sélectivité du récepteur semble être due à la petite taille de la poche de liaison pour la protéine G à l'intérieur de la cellule, alors que la protéine G stimulatrice nécessite un site de liaison plus grand.
Manglik a précédemment collaboré avec le laboratoire informatique de découverte de médicaments de Brian Shoichet, Doctorat., professeur de chimie pharmaceutique à l'École de pharmacie de l'UCSF, identifier une molécule appelée PZM21 qui permet au récepteur µ-opioïde d'engager uniquement la protéine G inhibitrice mais pas une autre molécule de signalisation appelée bêta-arrestine, et a montré que ce médicament sélectif soulageait la douleur avec des effets secondaires réduits chez la souris. Son laboratoire s'appuie maintenant sur le nouveau, portrait haute résolution du complexe récepteur opioïde - protéine G pour développer de nouveaux, composés encore plus sélectifs.