Mélanger la chimie computationnelle et les mathématiques théoriques s'est avéré une formule gagnante pour le chimiste d'Emory James Kindt (au centre), ses étudiants diplômés (de gauche à droite) Xiaokun Zhang et Lara Patel, et les étudiants diplômés en mathématiques Olivia Beckwith et Robert Schneider. Crédit :Stephen Nowland, Emory Photo/Vidéo
Les chimistes informaticiens et les mathématiciens ont développé une nouvelle méthode rapide pour calculer les constantes d'équilibre à l'aide de simulations à petite échelle, même lorsque la loi de l'action de masse ne s'applique pas.
Le Journal of Chemical Theory and Computation a publié l'algorithme et le logiciel résultants, que les chercheurs ont nommé PEACH – un acronyme pour « analyse par partition des histogrammes de clusters » et un clin d'œil au développement de la méthode en Géorgie à l'Université Emory.
« Notre méthode permettra aux chimistes informatiques de faire de meilleures prédictions dans les simulations pour un large éventail de réactions complexes, de la formation des aérosols dans l'atmosphère à la façon dont les protéines se réunissent pour former des filaments amyloïdes impliqués dans la maladie d'Alzheimer, " dit James Kindt, un professeur Emory de chimie computationnelle, dont le laboratoire a dirigé les travaux.
Auparavant, il fallait au moins une semaine de temps de calcul pour effectuer les calculs nécessaires à de telles prédictions. Le système PEACH réduit ce temps à quelques secondes en utilisant des astuces dérivées de la théorie des nombres.
"Notre outil peut utiliser un petit ensemble de données, puis extrapoler les résultats à un cas de grand système pour prédire la situation dans son ensemble, " dit Kindt.
"Ce qui a rendu ce projet si amusant et intéressant, ce sont ses aspects interculturels, " ajoute-t-il. " Les chimistes informaticiens et les mathématiciens théoriciens utilisent des langages différents et ne se parlent pas souvent. En travaillant ensemble, nous sommes tombés sur quelque chose qui semble être à la frontière des deux domaines."
L'équipe de recherche comprend Lara Patel et Xiaokun Zhang, qui sont tous deux doctorants. étudiants en chimie du laboratoire Kindt, et les théoriciens des nombres Olivia Beckwith et Robert Schneider, Emory Ph.D. candidats au Département de mathématiques et d'informatique. Chris Weeden, en tant que premier cycle Emory, contribué aux premières étapes des travaux.
La constante d'équilibre est un concept de base enseigné en première année de chimie collégiale. Selon la loi de l'action de masse, à une température donnée, quelle que soit la quantité de produit et de réactif mélangés, tant qu'ils sont à l'équilibre, un certain rapport produit/réactif sera égal à la constante d'équilibre.
"Cette équation est toujours vraie à l'équilibre pour un grand nombre de molécules, ", dit Kindt. "Peu importe qu'il soit appliqué à un seau d'eau ou à une seule goutte d'eau, qui se compose d'environ un milliard de milliards de molécules."
A des échelles beaucoup plus petites d'environ des dizaines de molécules, cependant, la loi de l'action de masse tombe en panne et ne s'applique pas.
Le laboratoire Kindt utilise des ordinateurs pour simuler le comportement des molécules, en particulier comment ils s'auto-assemblent en grappes. Octyl sulfate de sodium, ou SOS, est l'un des composés que le laboratoire utilise comme modèle expérimental. Le SOS est un tensioactif qui peut agir comme un détergent. Il forme de petits amas dans l'eau qui peuvent encapsuler l'huile et la graisse. Les simulations de la façon dont les molécules SOS se réunissent peuvent prédire la distribution des tailles d'amas formés dans différentes conditions, afin d'améliorer la conception des savons et des détergents, et mieux comprendre les processus biologiques tels que la façon dont les sels biliaires décomposent les globules de graisse au cours du processus digestif.
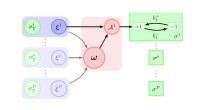
Dans un test clé de leur modèle, le laboratoire devait s'assurer que l'équilibre pour la réaction d'assemblage des molécules SOS en clusters correspondait aux expériences.
« Si nous devions effectuer des simulations avec un grand nombre de molécules, on pouvait compter les amas qui se formaient de chaque taille, compter les molécules restées libres des amas, et utiliser ces informations pour calculer la constante d'équilibre pour former chaque cluster de taille, " Dit Kindt. « Le défi auquel nous avons été confrontés était qu'il faudrait trop de temps aux ordinateurs pour effectuer des simulations d'un nombre suffisamment important de molécules pour que cela fonctionne. et pour le nombre de molécules de regroupement que nous pourrions pratiquement gérer – environ 50 – la loi de l'action de masse ne fonctionnerait pas. »
Kindt a décidé d'aborder le problème en considérant toutes les différentes manières dont les molécules dans une réaction pourraient se regrouper en grappes de différentes tailles afin d'arriver à une moyenne. Après avoir fait quelques lectures, il s'est rendu compte que ces différentes façons de regrouper les molécules étaient ce que les théoriciens des nombres appellent des partitions entières.
Une partition d'un nombre est une séquence d'entiers positifs qui s'additionnent à ce nombre. Par exemple, il y a cinq partitions du nombre 4 (4 =3+1 =2+2 =2+1+1 =1+1+1+1). Le nombre de partitions augmente à un rythme incroyable. Le nombre de partitions pour le nombre 10 est de 42. Pour le nombre 100, les cloisons explosent à plus de 190, 000, 000.
Cette même explosion de possibilités se produit pour les façons dont les molécules peuvent se regrouper.
Lara Patel et Xiaokun Zhang ont travaillé sur une méthode de « force brute » pour qu'un ordinateur parcoure toutes les manières de combiner 10 molécules d'un type avec 10 molécules d'un autre type. Le problème, c'est qu'il a fallu quelques jours à un ordinateur pour faire une seule analyse. Et le temps de calcul nécessaire si seulement quelques molécules supplémentaires étaient ajoutées à l'analyse augmentait de façon exponentielle.
Les chimistes informatiques s'étaient heurtés à un mur.
Kindt a contacté Ken Ono, un théoricien des nombres de renommée mondiale au département de mathématiques et d'informatique d'Emory, pour voir si l'un de ses étudiants diplômés serait intéressé à s'attaquer au problème.
Olivia Beckwith et Robert Schneider ont sauté sur l'occasion.
"Les simulations informatiques du laboratoire Kindt montrent que les théorèmes classiques de la théorie des partitions se produisent réellement dans la nature, même pour un petit nombre de molécules, " dit Schneider. " C'était surprenant et très cosmique pour moi d'apprendre que la théorie des nombres détermine les événements du monde réel. "
"C'était vraiment inattendu, " ajoute Beckwith. "En mathématiques théoriques, nous avons tendance à travailler indépendamment des phénomènes physiques comme l'interaction des molécules."
Les chimistes et les mathématiciens ont commencé à se rencontrer régulièrement pour discuter du problème et apprendre la terminologie de l'autre. "J'ai dû sortir le livre de chimie du lycée de mon fils et passer un week-end à le lire, " dit Schneider.
"C'est arrivé si organiquement, " dit Patel à propos du processus de mélange de leurs deux spécialités. "Olivia et Robert écrivaient des équations au tableau et dès qu'une formule avait un sens pour moi, je commençais à penser dans ma tête, « Comment pouvons-nous coder cela pour pouvoir l'appliquer ? »"
Les deux mathématiciens ont suggéré une stratégie qui pourrait rendre le problème beaucoup plus facile à calculer, basé sur un théorème connu sous le nom de formule de Faà di Bruno.
"C'était surprenant, " Zhang dit, « parce que c'était une idée qui ne me serait jamais venue à l'esprit. Ils nous ont aidés à nous décrocher et à trouver un moyen de faire avancer nos recherches. »
"Ils nous ont aidés à trouver un raccourci pour que nous n'ayons pas à générer toutes les partitions pour que les molécules puissent s'agglomérer, " Kindt ajoute. " Leur algorithme est un moyen beaucoup plus élégant et simple de trouver la moyenne globale. "
Patel et Zhang ont utilisé ce nouvel algorithme pour créer un logiciel permettant d'analyser les données des simulations informatiques. Le système résultant, PÊCHE, accélère les calculs qui prenaient auparavant de deux heures à une seule seconde. Après avoir démontré comment PEACH simplifie les simulations d'assemblages SOS, l'équipe de recherche s'apprête à simuler ce processus pour une gamme d'autres molécules.
"Nous sommes intéressés à décrire comment les structures moléculaires dictent l'assemblage dans tout type de scénario, tels que les premiers stades de la formation des cristaux, ", dit Kindt. "Nous travaillons également à quantifier l'endroit où la loi de l'action de masse s'effondre. Nous pourrions alors affiner la stratégie PEACH pour la rendre encore plus efficace."