Une nouvelle étude menée par des chercheurs de l'Université du Wisconsin-Madison a identifié la base structurelle de la façon dont les complexes protéiques étroitement liés sont brisés pour devenir inactivés. La structure explique pourquoi les complexes sont moins actifs dans certains cancers et maladies neurodégénératives, et offre un point de départ pour identifier des cibles médicamenteuses pour le réactiver.
Au fur et à mesure que nous grandissons, nos cellules répondent à des signaux étroitement régulés qui leur disent de croître et de se diviser jusqu'à ce qu'elles aient besoin de se développer en tissus et organes spécialisés. La plupart des cellules adultes sont spécialisées, et ils répondent correctement aux signaux qui leur disent d'arrêter de grandir. Les cancers peuvent se développer lorsque quelque chose ne va pas avec ces signaux.
Un de ces signaux « arrêter et se spécialiser » se trouve avec les complexes protéiques connus sous le nom de PP2A. Il existe environ 100 complexes PP2A connus, et ensemble, on estime qu'elles régulent près d'un tiers de toutes les protéines cellulaires. Ces complexes consistent en un noyau inactif jusqu'à ce qu'il se mélange et s'apparie avec l'une des nombreuses protéines de spécificité pour former des protéines étroitement liées, complexes PP2A actifs. Active PP2A utilise ces partenaires de spécificité pour trouver ses cibles - généralement des protéines pro-croissance - et les inactive. PP2A est un indice critique, alors, en contrôlant la croissance cellulaire et en maintenant des fonctions neurologiques normales. Sans surprise, il est muté dans de nombreux cancers et troubles neurologiques.
« Nous en savons beaucoup sur la formation des complexes PP2A actifs et identifient de plus en plus leurs cibles dans les cellules, mais nous savons très peu de choses sur la façon dont ils sont inactivés, " explique Yongna Xing, professeur agrégé d'oncologie au UW Carbone Cancer Center et au McArdle Laboratory for Cancer Research et auteur principal d'une nouvelle étude publiée aujourd'hui (22 décembre 2017) dans Communication Nature . "C'est un complexe très serré, c'est presque comme un rocher, mais il doit y avoir un moyen de le briser."
Les travaux antérieurs de Xing ont montré que la PP2A est inactive lorsqu'une protéine régulatrice, 4, est attaché. Cependant, lorsque les complexes PP2A actifs ont été testés avec 4, ils sont restés actifs, ce qui signifie qu'il devait y avoir un autre déclencheur qui a brisé le complexe.
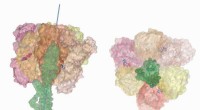
Dans la nouvelle étude, Xing et ses collègues identifient ce déclencheur comme la protéine TIPRL. Lorsqu'ils ont provoqué des complexes PP2A actifs avec 4 et TIPRL, les complexes se sont séparés. Prochain, ils ont déterminé la structure tridimensionnelle de TIRPL avec PP2A grâce à une technique connue sous le nom de cristallographie aux rayons X.
"La structure montre comment TIPRL peut attaquer les complexes PP2A actifs même s'il a une affinité beaucoup plus faible que les sous-unités de spécificité pour le noyau PP2A, " dit Xing. " Avec la structure, nous avons pu identifier comment TIRPL peut attaquer le complexe, changer sa conformation et, avec 4, le faire s'effondrer solidement. Il était difficile d'imaginer comment ce processus pourrait se produire sans connaissances structurelles. »
Si nous considérons le PP2A comme un tournevis électrique, les résultats ont beaucoup de sens pratique. La protéine centrale est la base motorisée, et les protéines de spécificité - celles qui se mélangent et s'assortissent pour aider PP2A à trouver la bonne cible - sont les têtes de vis. Lorsque vous souhaitez passer d'un tournevis cruciforme à un tournevis plat, vous ne jetez pas tout le complexe de tournevis électriques et n'en achetez pas un nouveau ; au lieu de cela, vous détachez une tête de vis et en fixez une autre. De la même manière, il est coûteux en énergie pour une cellule de dégrader l'ensemble du complexe PP2A, le rôle de TIPRL est donc de détacher la protéine de spécificité et de recycler le noyau PP2A.
L'une des découvertes les plus intéressantes de la structure était la façon dont la TIRPL flexible est comparée aux sous-unités de spécificité, incitant les chercheurs à se demander comment les mutations PP2A couramment observées chez les patients cancéreux affectent la liaison TIPRL. En utilisant un noyau normal ou PP2A contenant ces mutations, ils ont mesuré à quel point TIPRL et les sous-unités de spécificité peuvent s'y lier. Ils ont constaté que les mutations du noyau n'ont presque aucun effet sur la liaison TIPRL, mais ils affaiblissent considérablement la liaison des protéines de spécificité. Ces mutations, alors, probablement provoquer un passage des complexes PP2A actifs à la forme désassemblée et inactive.
« Dans de nombreuses maladies, y compris les cancers et les maladies neurodégénératives, PP2A en général est moins actif, souvent due à des mutations, " Xing note. " Cette structure aide à expliquer comment ces mutations conduisent à une régulation négative de PP2A en déplaçant l'équilibre vers la dissociation complexe induite par TIPRL. "
Avec la structure en main, Xing espère pouvoir mieux comprendre le cycle d'activation et d'inactivation de PP2A, et comment il régule la croissance cellulaire.
"Par exemple, la PP2A active est connue pour inhiber le K-ras, une protéine qui stimule la croissance dans de nombreuses tumeurs et n'a actuellement aucun bon inhibiteur clinique, " Xing dit. " Si vous pouvez trouver un moyen de réactiver PP2A, cela pourrait être très important dans le traitement de ces cancers."