Crédit :Laboratoire Ames
Le Critical Materials Institute du département de l'Énergie des États-Unis a mis au point un programme informatique, appelé ParFit, qui peuvent réduire considérablement le temps consacré à l'identification de composés chimiques prometteurs utilisés dans les méthodes de traitement des terres rares.
Tester et développer des méthodes plus efficaces et respectueuses de l'environnement pour extraire les métaux des terres rares aussi rapidement que possible est un objectif principal de CMI. Les métaux des terres rares sont essentiels à de nombreuses technologies énergétiques modernes, mais la forte demande commerciale et les défis miniers ont rendu l'optimisation de la production de notre pays et de leur utilisation d'une importance vitale.
"Traditionnel, les méthodes de mécanique quantique pour prédire la conception moléculaire et le comportement de ces extractants sont trop coûteuses en calculs, et prennent trop de temps pour l'échelle de temps nécessaire, " a déclaré Federico Zahariev, concepteur de logiciels et scientifique du CMI. " Nous avons donc développé un programme qui pourrait créer un modèle mécanique classique plus simple qui refléterait toujours la précision du modèle de mécanique quantique. "
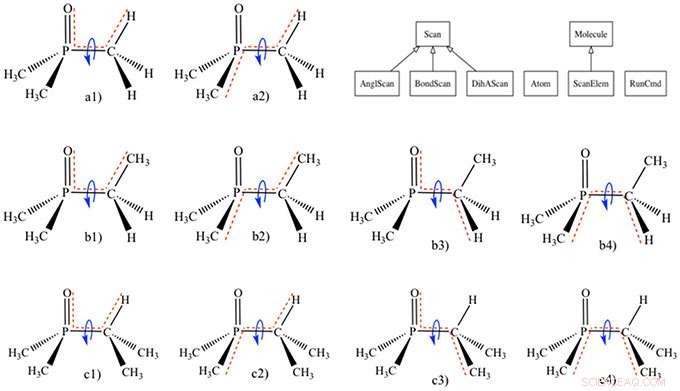
ParFit utilise des méthodes traditionnelles et avancées pour entraîner le modèle mécanique classique à ajuster les informations de mécanique quantique à partir d'un ensemble d'entraînement. Ces modèles classiques peuvent ensuite être utilisés pour prédire la forme de nouveaux extractants et la manière dont ils se lient aux métaux.
"Grosso modo, considérer la forme et la structure de la molécule comme un système de ressorts, où il peut être nécessaire de serrer ou desserrer les différentes connexions pour que cela fonctionne correctement, " a déclaré Theresa Windus, scientifique du CMI. " C'est de la même manière que nous appliquons les calculs de mécanique quantique pour créer ces modèles mécaniques classiques - c'est une tâche fastidieuse, sujet aux erreurs, et long processus. ParFit rend cela aussi rapide que possible, automatise l'ajustement de ces paramètres, et reproduit fidèlement les énergies de la mécanique quantique."
« Les capacités du programme permettent aux chercheurs de modéliser un nombre quasi illimité de nouveaux extractants, " a déclaré Marilu Dick-Perez, développeur de logiciels et scientifique CMI. Par exemple, les modèles classiques utilisés dans le code logiciel, HostDesigner - développé par Benjamin Hay du Supramolecular Design Institute, crée et évalue rapidement la viabilité des extractants possibles et cible les extractants les mieux adaptés à des recherches ultérieures. "Nous avons réduit le travail de calcul de 2-3 ans à trois mois, " a-t-elle déclaré. " Nous avons intégré autant de connaissances spécialisées que possible dans ce programme, afin que même un utilisateur novice puisse naviguer dans le programme."
Les capacités du logiciel sont discutées plus en détail dans un article, "ParFit :un programme orienté objet basé sur Python pour l'ajustement des paramètres de mécanique moléculaire aux données ab Initio", écrit par Federico Zahariev, Nuwan De Silva, Mark S. Gordon, Theresa L. Windus, et Marilu Dick-Perez, et publié dans le Journal of Chemical Information and Modelling .