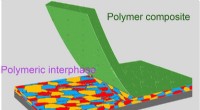
Vue d'artiste de molécules organiques s'adsorbant sur une surface de silicium. Crédit :Aaron Beller
Alors que de nouvelles méthodes sont devenues disponibles pour comprendre et manipuler la matière à ses niveaux les plus fondamentaux, les chercheurs travaillant dans le domaine interdisciplinaire de la science des matériaux ont de plus en plus réussi à synthétiser de nouveaux types de matériaux. Souvent, l'objectif des chercheurs dans le domaine est de concevoir des matériaux qui incorporent des propriétés qui peuvent être utiles pour exécuter des fonctions spécifiques. De tels matériaux peuvent, par exemple, être plus stable chimiquement ou résistant à la rupture physique, présentent des caractéristiques électromagnétiques avantageuses, ou réagir de manière prévisible à des conditions environnementales spécifiques.
Le Dr Ralf Tonner et son groupe de recherche à l'Université de Marburg relèvent le défi de la conception de matériaux fonctionnels d'une manière inhabituelle, en appliquant des approches basées sur la chimie computationnelle. Utilisation des ressources de calcul du High-Performance Computing Center Stuttgart (HLRS), l'un des trois centres nationaux allemands de calcul intensif qui composent le Gauss Center for Supercomputing, Tonner modélise des phénomènes qui se produisent à l'échelle atomique et subatomique pour comprendre comment des facteurs tels que la structure moléculaire, propriétés électroniques, une liaison chimique, et les interactions entre les atomes affectent le comportement d'un matériau.
"Quand vous étudiez comment, par exemple, une molécule s'adsorbe sur une surface, " Tonner explique, « d'autres scientifiques décriront souvent ce phénomène avec des méthodes de la physique, théorie de l'état solide, ou des structures de bande. Nous pensons qu'il peut également être très utile de demander, comment un chimiste regarderait-il ce qui se passe ici ?" De ce point de vue, Tonner cherche à savoir si la compréhension des réactions chimiques (comment les atomes se lient en molécules et réagissent lorsqu'ils sont mis en contact les uns avec les autres) peut offrir des perspectives nouvelles et utiles.
Dans une nouvelle publication en Sciences moléculaires computationnelles WIREs , Tonner et sa collaboratrice Lisa Pecher mettent en évidence la capacité des approches de chimie computationnelle utilisant le calcul haute performance à révéler des phénomènes intéressants qui se produisent entre les molécules organiques et les surfaces. Ils montrent aussi plus généralement comment ces interactions peuvent être appréhendées au regard du monde moléculaire et du solide. Les connaissances qu'ils ont acquises pourraient être utiles pour concevoir des surfaces à motifs, un objectif de scientifiques travaillant sur la prochaine génération de plus puissants, semi-conducteurs plus efficaces.
Apporter le calcul à la chimie
Les atomes se lient pour former des molécules et des composés lorsqu'ils se rapprochent, puis échangent ou partagent des électrons en orbite autour de leurs noyaux. Les atomes spécifiques impliqués, les formes physiques que prennent les molécules, leurs propriétés énergétiques, et comment ils interagissent avec d'autres molécules voisines sont toutes des propriétés qui donnent à un composé ses propriétés uniques. De telles caractéristiques peuvent déterminer si les composés sont susceptibles de rester stables, ou si des contraintes telles que des changements de température ou de pression pourraient affecter leur réactivité.
Tonner utilise une approche informatique appelée théorie de la fonctionnelle de la densité (DFT) pour explorer ces caractéristiques à l'échelle quantique; C'est, à l'échelle où la mécanique newtonienne est remplacée par le monde beaucoup plus étrange de la mécanique quantique (à des distances inférieures à 100 nanomètres). La DFT utilise des informations sur les variations de densité d'électrons au sein d'une molécule - une quantité qui peut également être mesurée expérimentalement à l'aide d'une technologie largement utilisée appelée diffraction des rayons X - pour dériver l'énergie du système. Cette, à son tour, permet aux chercheurs d'inférer les interactions entre les noyaux ainsi que les interactions entre les électrons et les noyaux, facteurs essentiels à la compréhension des liaisons et réactions chimiques.
DFT peut fournir utile, bien que statique, informations sur les profils énergétiques des composés qu'ils étudient. Pour mieux comprendre comment les systèmes de molécules se comportent réellement lorsqu'ils interagissent avec une surface, Le groupe de Tonner utilise également le calcul haute performance au HLRS pour effectuer des simulations de dynamique moléculaire. Ici, les scientifiques regardent comment le système de molécules se développe au fil du temps, au niveau des atomes et des électrons et à des échelles de temps de la picoseconde (une picoseconde équivaut à un trillionième de seconde).
De tels calculs utilisent généralement 2, 000-3, 000 cœurs de calcul, courir sur un problème pendant une semaine, et Tonner a été budgétisé à environ 30 millions d'heures CPU à HLRS pour le cycle de financement actuel de deux ans.

Vue d'artiste de molécules organiques s'adsorbant sur une surface de silicium. Crédit :Aaron Beller
"L'augmentation de la puissance de calcul a permis à la chimie computationnelle et à la chimie quantique de décrire de vrais systèmes moléculaires. Il y a à peine 15 à 20 ans, les gens ne pouvaient regarder que de petites molécules et devaient faire des approximations assez fortes, " explique Tonner. " Au cours des dernières années, les communautés de la chimie computationnelle et de la théorie de l'état solide ont résolu le problème de la parallélisation de leurs codes pour fonctionner efficacement sur des systèmes de calcul haute performance. Au fur et à mesure que les supercalculateurs grossissent, nous prévoyons être en mesure de développer des modèles de plus en plus réalistes pour les systèmes expérimentaux en science des matériaux."
Vers des semi-conducteurs à base de lumière
Un domaine dans lequel Tonner utilise actuellement la chimie informatique est d'étudier les moyens d'améliorer le silicium pour une utilisation dans de nouveaux types de semi-conducteurs. Ce problème est devenu urgent ces dernières années, car il est devenu clair que l'industrie de la microélectronique atteint les limites de sa capacité à améliorer les semi-conducteurs en utilisant uniquement du silicium.
Comme le rapportent Tonner et ses collègues expérimentaux dans un article récent du Beilstein Journal of Organic Chemisty, la fonctionnalisation du silicium avec des composés tels que le phosphure de gallium (GaP) ou l'arséniure de gallium (GaAs) pourrait permettre la conception de nouveaux types de semi-conducteurs. Cette recherche, basé dans un domaine appelé photonique sur silicium, postule que ces nouveaux matériaux permettraient d'utiliser la lumière à la place des électrons pour le transport du signal, soutenir le développement d'appareils électroniques améliorés.
"Pour faire ça, " Tonner explique, "Nous devons vraiment comprendre à quoi ressemblent et se comportent les interfaces entre le silicium et ces composés organiques. La réaction entre ces deux classes de matériaux doit se dérouler de manière très contrôlée afin que l'interface soit aussi parfaite que possible. Avec la chimie computationnelle, nous pouvons regarder aux détails élémentaires de ces interactions et processus."
Par exemple, recouvrir une plaque de silicium, des molécules précurseurs liquides des atomes constitutifs de l'arséniure de gallium sont placées dans un barboteur, où ils sont ensuite amenés en phase gazeuse. Ces molécules précurseurs sont composées des atomes nécessaires au nouveau matériau (gallium, arsenic) et des ions ou molécules appelés ligands pour les stabiliser en phase liquide et gazeuse. Ces ligands sont ensuite perdus dans le processus de dépôt et lorsque du silicium est placé dans le système, les molécules précurseurs sont adsorbées sur la surface solide de silicium. Après adsorption et perte des ligands, les atomes de gallium et d'arséniure se fixent au silicium, former un film de GaAs.
L'arrangement des atomes lorsqu'ils s'adsorbent sur une surface est déterminé par la liaison chimique. La force de ces liaisons et la densité avec laquelle les molécules précurseurs de GaAs sont adsorbées sont affectées non seulement par la distance entre elles et la surface du silicium mais également par les interactions entre les molécules précurseurs elles-mêmes. Dans un type d'interaction, appelé répulsion de Pauli, des nuages d'électrons se chevauchent et se repoussent, provoquant une baisse de l'énergie disponible pour la liaison. En autre, appelée interaction de dispersion attractive, les changements dans les positions électroniques dans un atome provoquent la redistribution des électrons dans d'autres atomes, harmoniser les mouvements des électrons et abaisser l'énergie de l'ensemble du système.
Précédemment, il a été suggéré que les relations répulsives entre les atomes sont le facteur le plus important pour « diriger » les atomes en place lorsqu'ils s'adsorbent sur une surface. En utilisant la théorie de la fonctionnelle de la densité et en examinant les caractéristiques intrigantes de la distribution des électrons, les chercheurs ont déterminé que la capacité des atomes à diriger d'autres atomes en place sur la surface peut également résulter d'interactions dispersives attrayantes.
Une meilleure compréhension de ces interactions fondamentales devrait aider les concepteurs de semi-conducteurs optiquement actifs à améliorer l'adsorption des molécules précurseurs sur le silicium. Cette, à son tour, permettrait de combiner la conduction du signal lumineux avec la microélectronique à base de silicium, réunissant le meilleur des deux mondes en conduction optique et électronique.
Pour Tonner, l'utilisation des méthodes des premiers principes en chimie pour les applications de la science des matériaux est très prometteuse. « La théorie est aujourd'hui très souvent prise en complément de l'investigation expérimentale, " dit-il. " Bien que l'expérimentation soit extrêmement importante, notre objectif ultime est que la théorie soit prédictive de manière à nous permettre de faire les premiers pas dans la conception de matériaux inspirés des premiers principes. Je vois cela comme un objectif à long terme."