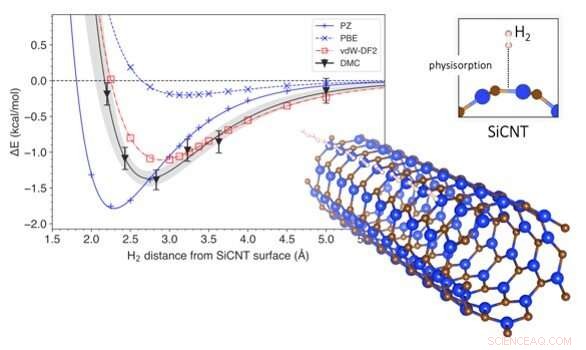
Le changement d'énergie associé à l'élimination de l'hydrogène des nanotubes de carbure de silicium. Le graphique montre la variation de l'énergie du système avec la distance d'une molécule d'hydrogène à la surface d'un nanotube de carbure de silicium (en bas à droite). La profondeur de la courbe signifie l'énergie nécessaire pour extraire l'hydrogène du stockage. Une comparaison des méthodes de prédiction est présentée, avec DMC étant le plus précis et vdW-DF2 étant son match le plus proche. Crédit :Kenta Hongo de JAIST
L'énergie hydrogène a le potentiel d'être une mesure clé pour atteindre l'objectif de zéro émission nette des Nations Unies, mais son utilisation industrielle a été entravée par la difficulté de son stockage et de sa manipulation. L'hydrogène devient un gaz à très basse température (-252°C), ce qui rend son stockage à température ambiante difficile. L'interaction entre l'hydrogène et son matériau de stockage est tout simplement trop faible pour persister à température ambiante. Cela rend la conception des matériaux de stockage cruciale pour atteindre l'objectif d'utilisation quotidienne de l'énergie hydrogène.
C'est là qu'intervient la conception informatique des matériaux. Beaucoup de temps et d'efforts peuvent être économisés lors du développement de la technologie de l'hydrogène en concevant un matériau sur un ordinateur et en simulant sa capacité de stockage d'hydrogène. Mais les prédictions deviennent très limitées dans leur utilisation à moins qu'elles ne soient exactes et puissent être faites à un coût de calcul raisonnable. Dans une étude récente publiée dans ACS Oméga , les scientifiques développent un calcul coûteux, nouvelle méthode mais très précise pour prédire le stockage de l'hydrogène :« L'amélioration de la fiabilité des prévisions pour les simulations peut aider à accélérer le développement de matériaux pour le stockage de l'hydrogène et conduire à une société plus économe en énergie, " déclare le Dr Kenta Hongo de l'Institut avancé japonais des sciences et de la technologie (JAIST), qui a dirigé l'étude.
L'une des forces d'attraction fondamentales entre les objets est la force de van der Waals, qui définit l'interaction entre des atomes ou des molécules en fonction de la distance qui les sépare. Puisque la force de Van der Waals est la conséquence de processus quantiques assez compliqués, les traitements conventionnels ne pouvaient pas bien le décrire, et donc les simulations jusqu'à présent sont au niveau des estimations approximatives de celui-ci. Mais est-il juste de le faire en simulant le stockage d'hydrogène ? C'était la principale préoccupation du Dr Hongo et de son équipe.
Pour répondre à cette question, ils se sont penchés sur les nanotubes de carbure de silicium, l'un des matériaux les plus prometteurs pour le stockage de l'hydrogène. En utilisant une technique de calcul appelée diffusion Monte Carlo (DMC), ils ont créé un modèle qui tenait compte des forces de van der Waals lors de la simulation du stockage de l'hydrogène dans des nanotubes de carbure de silicium. La plupart des modèles conventionnels considèrent les interactions entre l'hydrogène et les nanotubes de carbure de silicium dans leur ensemble, mais la méthode DMC utilise la puissance d'un supercalculateur pour reconstituer fidèlement le mécanisme d'interaction en suivant l'arrangement des électrons individuels. Cela fait du modèle DMC la méthode de prédiction la plus précise à ce jour. En utilisant le modèle DMC, les chercheurs ont également pu prédire la quantité d'énergie nécessaire pour déloger l'hydrogène de son stockage, et à quelle distance l'hydrogène était susceptible d'être de la surface du nanotube de carbure de silicium. Ils ont ensuite comparé les résultats de leur modélisation à ceux obtenus via des méthodes de prédiction conventionnelles.
Les méthodes de prédiction conventionnelles sont généralement basées sur des techniques de calcul appelées théorie de la fonctionnelle de la densité (DFT). La DFT utilise des fonctionnelles (descriptions de modèles d'interactions quantiques) qui décrivent les variations spatiales de la densité électronique pour déterminer les propriétés des systèmes complexes. Bien qu'il y ait eu plusieurs études DFT sur le stockage de l'hydrogène sur des nanotubes de carbure de silicium, aucun d'entre eux n'a incorporé les forces de van der Waals dans ses prédictions. Les fonctionnelles DFT corrigées par Van der Waals ont, cependant, été utilisé dans la prédiction d'autres matériaux. Le Dr Hongo et son équipe ont simulé le stockage d'hydrogène à l'aide d'un large éventail de fonctions DFT, ceux avec des corrections van der Waals et ceux sans. Ils ont constaté que les fonctionnelles DFT sans les corrections de van der Waals sous-estimaient l'énergie requise pour le stockage de l'hydrogène de 4 à 14 %. D'autre part, Les fonctionnelles DFT corrigées par van der Waals ont produit des résultats assez similaires à ceux du DMC. De plus, ils ont constaté que la contribution de la force de van der Waals à l'énergie de stockage était d'environ 9-29%, ce qui n'est guère négligeable.
Ces découvertes, Le Dr Hongo croit, peut être un tremplin pour de nouvelles innovations dans la technologie de simulation du stockage de l'hydrogène. "Bien que la méthode DMC soit coûteuse en calcul, il peut être utilisé pour clarifier les particularités (tendances d'erreur de prédiction) de chaque méthode de prédiction. Cela nous aidera à comprendre à quelle prédiction faire confiance, et aussi comment modifier les méthodes de prédiction pour les rendre plus utiles, " il explique.