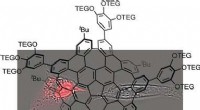
L'apprentissage automatique prédit la structure et la dynamique des nanoparticules Des nanostructures telles que ces nanoparticules d'or recouvertes de thiol peuvent désormais être étudiées en utilisant la nouvelle méthode d'apprentissage automatique développée à l'Université de Jyväskylä. La méthode peut prédire de manière fiable l'énergie potentielle d'une structure donnée. Crédit :Antti Pihlajamäki/Université de Jyväskylä
Des chercheurs du Centre des nanosciences et de la Faculté des technologies de l'information de l'Université de Jyväskylä en Finlande ont démontré que les nouvelles méthodes d'apprentissage automatique à distance développées à l'Université de Jyväskylä sont capables de prédire de manière fiable les structures et la dynamique atomique des nanoparticules. Les nouvelles méthodes sont nettement plus rapides que les méthodes de simulation traditionnelles utilisées pour la recherche sur les nanoparticules et faciliteront des explorations plus efficaces des réactions particule-particule et de la fonctionnalité des particules dans leur environnement. L'étude a été publiée dans un numéro spécial consacré à l'apprentissage automatique dans le Journal de chimie physique le 15 mai, 2020.
Les nouvelles méthodes ont été appliquées à des nanoparticules métalliques stabilisées par un ligand, qui ont été longtemps étudiés au Nanoscience Center de l'Université de Jyväskylä. L'année dernière, les chercheurs ont publié une méthode capable de prédire avec succès les sites de liaison des molécules de ligand stabilisant à la surface des nanoparticules. Maintenant, un nouvel outil a été créé qui peut prédire de manière fiable l'énergie potentielle en fonction de la structure atomique de la particule, sans avoir besoin d'utiliser des calculs de structure électronique numériquement lourds. L'outil facilite les simulations Monte Carlo de la dynamique atomique des particules à des températures élevées.
L'énergie potentielle d'un système est une grandeur fondamentale en nanoscience computationnelle, puisqu'il permet des évaluations quantitatives de la stabilité du système, taux de réactions chimiques et forces des liaisons interatomiques. Les nanoparticules métalliques stabilisées par un ligand ont de nombreux types de liaisons interatomiques de force chimique variable, et traditionnellement, les évaluations énergétiques ont été effectuées en utilisant la théorie de la fonctionnelle de la densité (DFT) qui aboutit souvent à des calculs numériquement lourds nécessitant l'utilisation de supercalculateurs. Cela a empêché des simulations efficaces pour comprendre les fonctionnalités des nanoparticules, par exemple., comme catalyseurs, ou des interactions avec des objets biologiques tels que des protéines, virus, ou ADN. Méthodes d'apprentissage automatique, une fois formé pour modéliser les systèmes de manière fiable, peut accélérer les simulations de plusieurs ordres de grandeur.
La nouvelle méthode a permis d'exécuter des simulations sur un ordinateur portable ou un ordinateur de bureau
Dans ce travail, les chercheurs ont utilisé les énergies potentielles, prédit par la méthode d'apprentissage automatique, pour simuler la dynamique atomique de nanoparticules d'or stabilisées au thiol. Les résultats étaient en bon accord avec les simulations effectuées en utilisant la théorie de la fonctionnelle de la densité. La nouvelle méthode permettait d'exécuter des simulations sur un ordinateur portable ou un ordinateur de bureau sur une échelle de temps de quelques heures, tandis que les simulations DFT de référence prenaient des jours dans un superordinateur et utilisaient simultanément des centaines, voire des milliers de cœurs d'ordinateur. L'accélération permettra des simulations à long terme des changements structurels des particules et des réactions particule-particule à des températures élevées.
Les chercheurs ont utilisé une méthode d'apprentissage automatique à distance développée dans le groupe du professeur Tommi Kärkkäinen à Jyväskylä. Il décrit chaque configuration atomique momentanée d'une nanoparticule en calculant un soi-disant descripteur, et compare les distances entre les descripteurs dans un espace numérique multidimensionnel. En utilisant des corrélations avec un ensemble d'apprentissage créé par les simulations DFT de référence, l'énergie potentielle peut être prédite. Cette approche, utilisé maintenant pour la première fois dans la recherche sur les nanoparticules, est plus simple et plus transparent que les réseaux de neurones traditionnellement utilisés.
« Il est extrêmement motivant de pouvoir réduire la charge de calcul de l'exécution de simulations dans des superordinateurs à leur exécution avec une qualité similaire sur un ordinateur portable ou un PC domestique, ", déclare Antti Pihlajamäki, étudiant au doctorat, auteur principal de l'étude.
« Ce fut une grande surprise que nos méthodes d'apprentissage automatique relativement simples fonctionnent si bien pour les nanostructures complexes, " déclare le professeur Tommi Kärkkäinen.
« Dans la phase suivante, notre objectif est de généraliser la méthode pour qu'elle fonctionne bien pour des nanoparticules de différentes tailles et compositions chimiques. Nous aurons toujours besoin de supercalculateurs pour générer suffisamment de données de haute qualité pour entraîner l'algorithme d'apprentissage automatique, mais nous espérons qu'à l'avenir, nous pourrons utiliser ces nouvelles méthodes principalement pour étudier la fonctionnalité des nanoparticules dans des environnements chimiques complexes, " résume le professeur de l'Académie Hannu Häkkinen, qui a coordonné l'étude.