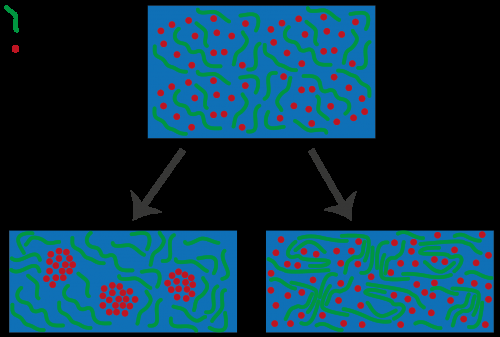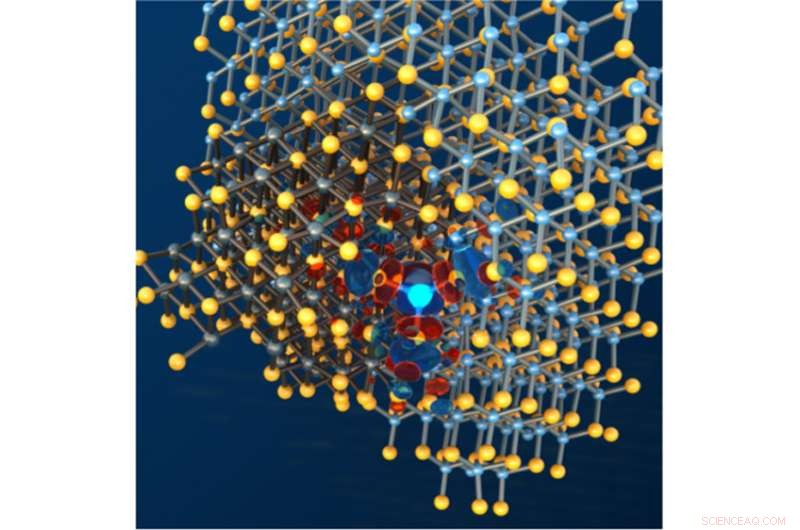
Coupe transversale de l'interface entre une nanoparticule de chalcogénure de plomb et sa matrice de chalcogénure de cadmium enrobée. Lorsqu'il est intégré dans des dispositifs optoélectroniques, il suffit d'avoir un seul atome au mauvais endroit à l'interface (représenté par la couleur bleu brillant) pour compromettre leurs performances. Crédit :Peter Allen, Institut de génie moléculaire, Université de Chicago
Pour comprendre la nature de quelque chose d'extrêmement complexe, vous devez souvent étudier ses plus petites parties. En essayant de déchiffrer l'univers, par exemple, nous recherchons des ondes gravitationnelles ou de faibles ondes lumineuses du Big Bang. Et pour comprendre l'essence même de la matière elle-même, nous le décomposons au niveau subatomique et utilisons des simulations informatiques pour étudier des particules comme les quarks et les gluons.
Comprendre les matériaux avec des fonctions spécifiques, tels que ceux utilisés dans les cellules solaires, et les moyens d'ingénierie pour améliorer leurs propriétés posent bon nombre des mêmes défis. Dans l'effort continu pour améliorer l'efficacité de conversion d'énergie des cellules solaires, les chercheurs ont commencé à creuser plus profondément, dans certains cas jusqu'au niveau atomique, pour identifier les défauts matériels qui peuvent nuire au processus de conversion.
Par exemple, les matériaux nanostructurés hétérogènes sont largement utilisés dans une variété de dispositifs optoélectroniques, y compris les cellules solaires. Cependant, en raison de leur nature hétérogène, ces matériaux contiennent des interfaces nanométriques présentant des défauts structurels pouvant affecter les performances de ces dispositifs. Il est très difficile d'identifier ces défauts dans les expériences, ainsi, une équipe de chercheurs du Laboratoire national d'Argonne du Département de l'énergie et de l'Université de Chicago a décidé d'exécuter une série de calculs atomistiques au Centre national de calcul scientifique de la recherche énergétique (NERSC) du Lawrence Berkeley National Laboratory pour trouver la cause première des défauts dans deux matériaux semi-conducteurs utilisés – séléniure de plomb (PbSe) et séléniure de cadmium (CdSe) – et fournissent des règles de conception pour les éviter.
« Nous sommes intéressés à comprendre les points quantiques et les nanostructures et leurs performances pour les cellules solaires, " dit Giulia Galli, Liew Family professeur de génie moléculaire à l'Université de Chicago et co-auteur d'un article publié dans Lettres nano qui présente ce travail et ses conclusions. "Nous faisons du mannequinat, en utilisant à la fois la dynamique moléculaire classique et les méthodes du premier principe, pour comprendre la structure et les propriétés optiques de ces nanoparticules et points quantiques."
Nanoparticules cœur-coquille
Pour cette étude, l'équipe s'est concentrée sur les nanoparticules hétérostructurées, dans ce cas une boîte quantique colloïdale dans laquelle des nanoparticules de PbSe sont noyées dans du CdSe. Ce type de point quantique, également connu sous le nom de nanoparticule cœur-coquille, ressemble à un œuf, Marton Vörös, Aneesur Rahman Fellow à Argonne et co-auteur de l'article, expliqué, avec un "jaune" fait d'un matériau entouré d'une "coquille" faite de l'autre matériau.
« Des expériences ont suggéré que ces nanoparticules hétérostructurées sont très favorables à la conversion de l'énergie solaire et aux transistors à couches minces, " a déclaré Vörös.
Par exemple, tandis que les efficacités de conversion d'énergie des points quantiques colloïdaux oscillent actuellement autour de 12% en laboratoire, « nous visons à prédire que les modèles structuraux de points quantiques dépasseront les 12 %, " a déclaré Federico Giberti, chercheur postdoctoral à l'Institute for Molecular Engineering de l'Université de Chicago et premier auteur sur le Lettres nano papier. « Si 20 % d'efficacité pouvaient être atteints, nous aurions alors un matériau qui devient intéressant pour la commercialisation. "
Pour que cela se produise, cependant, Vörös et Giberti ont réalisé qu'ils devaient mieux comprendre la structure des interfaces à l'échelle nanométrique et si des défauts atomistiques étaient présents. Donc, avec Galli, ils ont développé une stratégie de calcul pour étudier, au niveau atomique, l'effet de la structure des interfaces sur les propriétés optoélectroniques des matériaux. En utilisant la dynamique moléculaire classique et les méthodes des premiers principes qui ne reposent sur aucun paramètre ajusté, leur cadre leur a permis de construire des modèles informatiques de ces points quantiques intégrés.
En utilisant ce modèle comme base pour une série de simulations exécutées au NERSC, l'équipe de recherche a pu caractériser les points quantiques PbSe/CdSe et a découvert que les atomes qui sont déplacés à l'interface et leurs états électroniques correspondants - ce qu'ils appellent des "états pièges" - peuvent compromettre les performances des cellules solaires, expliqua Giberti. Ils ont ensuite pu utiliser le modèle pour prédire un nouveau matériau qui n'a pas ces états de piège et qui devrait mieux fonctionner dans les cellules solaires.
« En utilisant notre cadre de calcul, nous avons également trouvé un moyen d'ajuster les propriétés optiques du matériau en appliquant une pression, " ajouta Giberti.
Cette recherche, qui comprenait des études sur les structures électroniques et atomiques, a utilisé quatre millions d'heures de calcul intensif au NERSC, selon Vörös. La plupart des calculs de structure atomique ont été exécutés sur Cori, Le système 30 pétaflops de la NERSC installé en 2016, bien qu'ils aient également utilisé le système Edison, un Cray XC30 avec processeurs Intel Xeon. Bien que les calculs n'aient pas nécessité un grand nombre de processeurs, Giberti a noté, "J'avais besoin de lancer plusieurs simulations simultanées en même temps, et analyser toutes les données était en soi une tâche plutôt difficile."
Regarder vers l'avant, l'équipe de recherche prévoit d'utiliser ce nouveau cadre de calcul pour étudier d'autres matériaux et structures.
"Nous pensons que nos modèles atomistiques, lorsqu'il est associé à des expériences, apportera un outil prédictif pour les matériaux nanostructurés hétérogènes qui peuvent être utilisés pour une variété de systèmes semi-conducteurs, " Federico a déclaré. "Nous sommes très enthousiasmés par l'impact possible de notre travail."