Certains types de molécules forment des motifs lorsqu'ils sont déposés sur des substrats. Les dispositifs photovoltaïques et capteurs à partir de composés organiques dépendent de ce phénomène d'auto-organisation. Physiciens de la Ludwig-Maximilians-Universitaet à Munich, Allemagne, ont maintenant développé un modèle qui prédit ces modèles et permet ainsi l'optimisation de la synthèse moléculaire à l'avenir.
Certaines classes de molécules sont capables de s'organiser selon des motifs spécifiques sur des surfaces. Cette capacité d'auto-organisation est cruciale pour de nombreuses applications technologiques, qui dépendent de l'assemblage de structures ordonnées sur des surfaces. Cependant, il a été jusqu'à présent pratiquement impossible de prédire ou de contrôler le résultat de tels processus.
Maintenant, un groupe de chercheurs dirigé par le Dr Bianca Hermann, un physicien du Center for Nanoscience (CeNS) du LMU Munich, rapporte une percée significative :en combinant la physique statistique et des simulations détaillées avec des images obtenues par microscopie à effet tunnel (STM), l'équipe a pu formuler un modèle simple qui peut prédire les modèles observés. "Avec l'aide du modèle, nous pouvons générer une grande variété de motifs qui reproduisent étonnamment bien les arrangements observés expérimentalement", dit Hermann. "Nous voulons étendre cette approche à d'autres symétries de surface. Déjà maintenant les domaines de l'électronique moléculaire, applications de capteurs, la catalyse de surface et le photovoltaïque organique peuvent bénéficier de notre modèle. Sa capacité à prédire les structures formées par l'auto-organisation permet d'optimiser les briques moléculaires avant la synthèse." ( Lettres nano en ligne, 16 février 2010)
Quand « mère nature » fait l'ingénierie, les molécules peuvent s'auto-organiser en structures complexes - une première étape dans la formation de membranes, cellules et autres systèmes moléculaires. Le principe de l'auto-organisation, ce qui permet une utilisation très économe des ressources, est également exploitée dans la production de surfaces fonctionnalisées nécessaires à l'électronique moléculaire, applications de capteurs, catalyse et composants photovoltaïques. L'idée du processus de fabrication est que les composants moléculaires sont mis en contact avec un matériau de substrat, puis "par magie" trouvent leurs positions préférées dans le réseau moléculaire souhaité. Les composants de départ sont sélectionnés pour présenter des caractéristiques structurelles et chimiques spécifiques destinées à l'application envisagée. Cependant, l'optimisation des adlayers moléculaires dépend en grande partie d'une approche essai-erreur, et est donc compliqué et prend du temps.
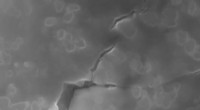
Pour développer le nouveau modèle de site d'interaction moléculaire, Le groupe du Dr Herrmann a collaboré avec Priv. Doz. Dr. Thomas Franosch et professeur Erwin Frey au sein du pôle d'excellence "Nanosystems Initiative Munich" (NIM). Le problème a été abordé en utilisant une approche de physique statistique connue sous le nom de méthode de Monte Carlo, ce qui permet d'effectuer une simulation informatique détaillée sur les statistiques des interactions moléculaires. Les motifs structuraux ainsi générés ont été comparés à des images expérimentales à haute résolution de motifs moléculaires obtenus par STM. Marta Balbas Gambra, un doctorant, a commencé chaque simulation avec une représentation mathématique d'une collection de centaines de particules orientées au hasard de conformation définie. Ces molécules schématiques ont ensuite été perturbées par - informatiquement - en ajoutant de l'énergie, obligeant la population à adopter une nouvelle configuration.
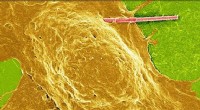
En utilisant cette stratégie de simulation, on peut générer une plus grande variété de motifs que ceux que l'on trouve naturellement, et beaucoup d'entre eux correspondaient étroitement aux modèles moléculaires réels révélés par STM. "Dans un cas, nous avons en fait prédit un modèle qui n'a été vérifié que plus tard avec STM", rapporte le doctorant Carsten Rohr. Selon les lois de la thermodynamique, les systèmes physiques ont tendance à adopter l'état avec l'énergie la plus favorable (c'est-à-dire la plus faible). Des tests expérimentaux ont montré que différentes configurations moléculaires s'interconvertissent jusqu'à ce qu'un arrangement prédomine qui rappelle les traces de pneus. Et en effet, l'approche de Monte Carlo avait prédit que cet arrangement correspond à l'état ayant la plus faible énergie.
"À la fin, nous avons pu montrer que la géométrie moléculaire et quelques traits saillants codent les motifs structuraux observés", explique le théoricien François. "Nous envisageons d'étendre l'approche à d'autres types de symétries de surface, mais le modèle fournit déjà un outil théorique important, car il nous aide à prévoir le type de motif de surface que formera une molécule fonctionnelle donnée. Cela signifie que la conception des molécules peut être optimisée pendant la phase de synthèse, afin d'obtenir des surfaces aux caractéristiques souhaitées", dit Hermann. Les physiciens du groupe, qui viennent d'horizons scientifiques différents et ont pu mettre en commun leur expertise pour ce projet, envisager de multiples applications potentielles de leur modèle en électronique moléculaire, technologie des capteurs, catalyse et photovoltaïque. D'autres possibilités incluent son utilisation pour prédire les résultats d'autres types d'interactions moléculaires également sur des substrats partiellement modelés.