Des chercheurs de la Pritzker School of Molecular Engineering (PME) de l'Université de Chicago, du Argonne National Laboratory et de l'Université de Modène et de Reggio Emilia ont développé un nouvel outil informatique pour décrire le comportement des atomes contenus dans les matériaux quantiques lorsqu'ils absorbent et émettent de la lumière. /P>
L'outil sera publié dans le cadre du progiciel open source WEST, développé au sein du Midwest Integrated Center for Computational Materials (MICCoM) par une équipe dirigée par le professeur Marco Govoni, et il aide les scientifiques à mieux comprendre et concevoir de nouveaux matériaux pour l'informatique quantique. technologies.
"Ce que nous avons fait, c'est élargir la capacité des scientifiques à étudier ces matériaux pour les technologies quantiques", a déclaré Giulia Galli, professeur de génie moléculaire de la famille Liew et auteur principal de l'article, publié dans Journal of Chemical Theory and Computation . "Nous pouvons désormais étudier des systèmes et des propriétés qui n'étaient vraiment pas accessibles, à grande échelle, dans le passé."
Le groupe de Galli a montré la précision de l'outil, connu sous le nom de WEST-TDDFT (Without Empty States—Time-Dependent Density Functional Theory), dans l'étude de trois matériaux différents à base de semi-conducteurs, mais a déclaré qu'il pouvait être appliqué à un large éventail de matériaux connexes et que le les logiciels développés peuvent fonctionner à grande échelle sur plusieurs architectures hautes performances.
Les unités d’information fondamentales qui sous-tendent les nouvelles technologies quantiques puissantes sont les qubits. Contrairement aux bits utilisés en informatique classique, qui utilisent uniquement des 0 et des 1 pour coder les données, les qubits peuvent également exister dans des états de superposition, représentant à la fois 0 et 1 simultanément.
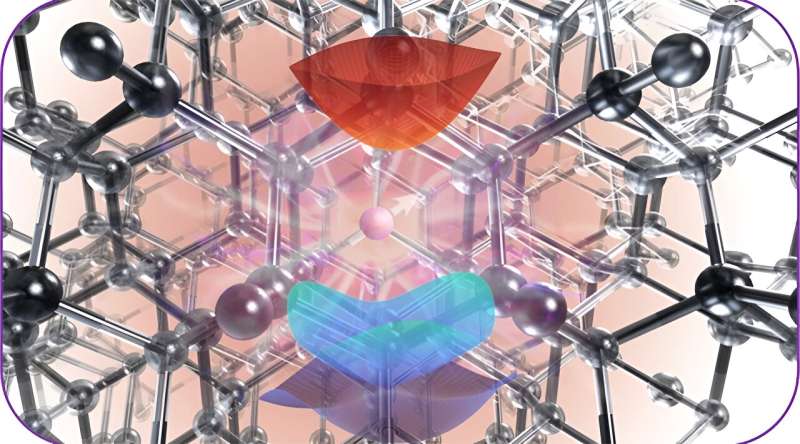
De minuscules défauts dans les matériaux, comme un atome manquant ou substitué dans le réseau structuré d'un cristal, peuvent prendre des états quantiques et être utilisés comme qubits. Ces qubits sont extrêmement sensibles aux propriétés électriques, optiques et magnétiques de leur environnement, ce qui leur donne la possibilité d'être utilisés comme capteurs.
Comprendre exactement comment ces « défauts ponctuels » interagissent avec les photons de lumière pour modifier leurs états énergétiques peut permettre aux chercheurs de mieux les manipuler ou de concevoir des matériaux qui utilisent les qubits comme capteurs ou unités de stockage de données.
"La manière dont ces matériaux absorbent et émettent de la lumière est essentielle pour comprendre comment ils fonctionnent pour les applications quantiques", a déclaré Galli. "La lumière est la façon dont vous interrogez ces matériaux."
Jusqu'à présent, les chercheurs pouvaient prédire à la fois l'absorption et l'émission de lumière par défauts ponctuels, mais ne pouvaient pas expliquer complètement certains des processus atomiques qui se produisaient dans le matériau lorsqu'il était dans son état excité, en particulier dans le cas de systèmes vastes et complexes. /P>
Les équations de mécanique quantique qui doivent être résolues pour déterminer les propriétés atomiques des matériaux sont incroyablement complexes et nécessitent une grande puissance de calcul. Dans ce nouveau travail, l'équipe de Galli a codé une nouvelle façon de résoudre de telles équations plus efficacement que par le passé tout en prouvant qu'elles étaient toujours exactes.
La rapidité et l'efficacité accrues avec lesquelles les équations peuvent désormais être résolues signifient qu'elles peuvent être appliquées plus facilement à des systèmes plus vastes. Dans le passé, le temps et la puissance de calcul requis pour analyser ces systèmes rendaient cela irréalisable.
"Grâce à ces méthodes, nous pouvons étudier l'interaction de la lumière avec les matériaux dans des systèmes assez vastes, ce qui signifie que ces systèmes sont plus proches des systèmes expérimentaux réellement utilisés en laboratoire", a déclaré Yu Jin, étudiant diplômé, premier auteur de l'étude. nouveau papier.
L'approche efficace développée par l'équipe peut fonctionner sur deux architectures informatiques différentes :les unités centrales de traitement (CPU) et les unités de traitement graphique (GPU). Les chercheurs l'ont utilisé pour étudier les propriétés de l'état excité des défauts ponctuels dans trois matériaux :le diamant, le carbure de silicium 4H et l'oxyde de magnésium. Ils ont découvert que l'outil pouvait calculer efficacement les propriétés de ces systèmes même lorsqu'ils contenaient des centaines ou des milliers d'atomes.
L'équipe MICCoM développant WEST comprend le Dr Victor Yu, Yu Jin et le professeur Marco Govoni. Le groupe continue d'appliquer et d'affiner les algorithmes disponibles dans le package, notamment WEST-TDDFT, pour étudier de larges classes de matériaux, non seulement pour les technologies quantiques, mais également pour les applications à faible consommation et énergie.
"Nous avons trouvé un moyen de résoudre plus efficacement les équations décrivant l'émission et l'absorption de la lumière afin qu'elles puissent être appliquées à des systèmes réalistes", a déclaré Govoni. "Nous avons montré que la méthode est à la fois efficace et précise."
Le nouvel outil s’inscrit dans l’objectif plus large du laboratoire Galli d’étudier et de concevoir de nouveaux matériaux quantiques. En outre, ce mois-ci, ils ont publié de nouveaux résultats montrant comment les défauts de spin proches de la surface d'un matériau se comportent différemment de ceux plus profonds à l'intérieur d'un matériau, en fonction de la manière dont la surface est terminée. Leurs résultats ont des implications pour la conception de capteurs quantiques reposant sur des défauts de spin.
L'équipe a également publié un article récent dans npj Computational Materials. , examinant les propriétés des matériaux ferroélectriques utilisés en informatique neuromorphique.
Plus d'informations : Yu Jin et al, Propriétés d'état excité des défauts ponctuels dans les semi-conducteurs et les isolants étudiées avec la théorie fonctionnelle de la densité dépendant du temps, Journal of Chemical Theory and Computation (2023). DOI :10.1021/acs.jctc.3c00986
Fourni par l'Université de Chicago