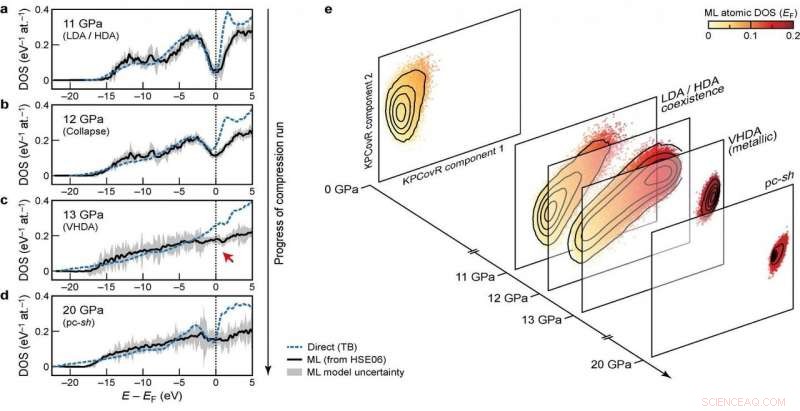
Densités d'états électroniques (DOS) à différentes étapes du cycle de compression Crédit :@Michele Ceriotti
La combinaison de calculs de structure électronique et de techniques d'apprentissage automatique (ML) est devenue une approche courante dans la modélisation atomistique de la matière. L'utilisation conjointe des deux techniques a permis aux chercheurs, par exemple, pour créer des modèles qui utilisent les coordonnées atomiques comme seules entrées pour prédire à moindre coût toute propriété pouvant être calculée par les calculs des premiers principes qui avaient été utilisés pour les entraîner.
Alors que les efforts les plus anciens et les plus avancés se sont concentrés sur l'utilisation des prédictions des énergies totales et des forces atomiques pour construire des potentiels interatomiques, des efforts plus récents ont ciblé des propriétés supplémentaires des cristaux et des molécules telles que les énergies d'ionisation, blindages chimiques RMN, propriétés de réponse diélectrique et densité de charge. Dans l'article "Apprendre la densité électronique des états dans la matière condensée, " Ceriotti et ses collègues se concentrent sur la densité d'états électronique (DOS), une autre quantité qui sous-tend de nombreuses propriétés de matériaux utiles, dont certains peuvent être observés expérimentalement.
Le DOS est essentiellement le nombre d'états différents que les électrons peuvent occuper à un niveau d'énergie particulier, et peut être utilisé, par exemple, calculer la contribution électronique à la capacité calorifique des métaux et la densité des porteurs de charges libres dans les semi-conducteurs. Il s'agit d'une approximation indirecte de propriétés telles que la bande interdite énergétique, l'énergie de la bande et le spectre d'absorption optique.
"La prédiction du DOS est un exercice intéressant en soi car c'est essentiellement la description la plus simple possible de la structure électronique au-delà de l'image de l'état fondamental, " a déclaré Ceriotti. " C'est aussi utile car il y a beaucoup de propriétés que vous pouvez calculer à partir du DOS, ce qui en fait un excellent exemple de la façon dont la prochaine génération de modèles ML peut être utilisée de la même manière que les calculs de structure électronique, en les utilisant de manière indirecte pour calculer des quantités intermédiaires qui peuvent ensuite être facilement traitées pour évaluer des propriétés plus difficiles à apprendre directement."
En développant le modèle, le groupe a cherché à assurer la transférabilité à travers différentes phases ainsi que l'évolutivité vers de grandes tailles de système. Leur approche ultime, qui examine comment différentes configurations atomiques affectent la distribution des niveaux d'énergie, atteint ces objectifs :il a pu apprendre et prédire le DOS calculé par DFT pour un ensemble de données diversifié de structures de silicium, couvrant un large éventail de conditions thermodynamiques et de différentes phases. Il évolue également de manière linéaire, plutôt qu'avec le cube du nombre d'atomes comme avec les calculs de structure électronique, ce qui la rend applicable aux grandes structures. Finalement, le modèle a permis une analyse du DOS local, donnant aux chercheurs la possibilité d'examiner l'interaction entre les motifs structurels et la structure électronique.
La combinaison de la transférabilité, et l'évolutivité des prédictions à de grandes tailles de système, rendre le modèle applicable pour répondre à des questions ouvertes de longue date en science des matériaux. Le nouveau cadre a déjà été utilisé pour élucider les propriétés électroniques d'une simulation à 100'000 atomes de silicium amorphe, subissant une série de transitions de phase lorsqu'il est comprimé à 20 Gpa, dans un article publié dans La nature aujourd'hui en collaboration avec une équipe composée de chercheurs d'Oxford, Cambridge, l'US Naval Research Laboratory et l'Université de l'Ohio. Le DOS prédit est également utilisé pour expliquer comment les transformations structurelles induites par la pression se couplent à la structure électronique du matériau.
La combinaison du nouveau modèle avec l'un des modèles d'énergie potentielle bien établis permet également de calculer les contributions électroniques aux propriétés macroscopiques telles que la capacité thermique des métaux et d'effectuer des simulations qui prennent en compte les effets de la température électronique finie - comme démontré dans un autre article à paraître prochainement sur les propriétés à haute température du nickel. En effet, le nouveau modèle est une étape critique vers l'objectif de MARVEL de développer des modèles d'apprentissage automatique intégrés qui augmentent - et peut-être éventuellement remplacent - les calculs de structure électronique coûteux.
"Il y a d'autres propriétés en dehors de la densité électronique des états, telles que les excitations optiques, et réponse RMN, que nous avons pu prédire avec précision grâce à l'apprentissage automatique. » a déclaré Ceriotti. « Si nous pouvons tous les utiliser en combinaison avec des potentiels interatomiques bon marché et précis, cela nous permettra de décrire toutes les propriétés des matériaux avec la même précision que celle obtenue avec l'électronique. calcul de structure, mais à une infime fraction du coût."