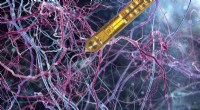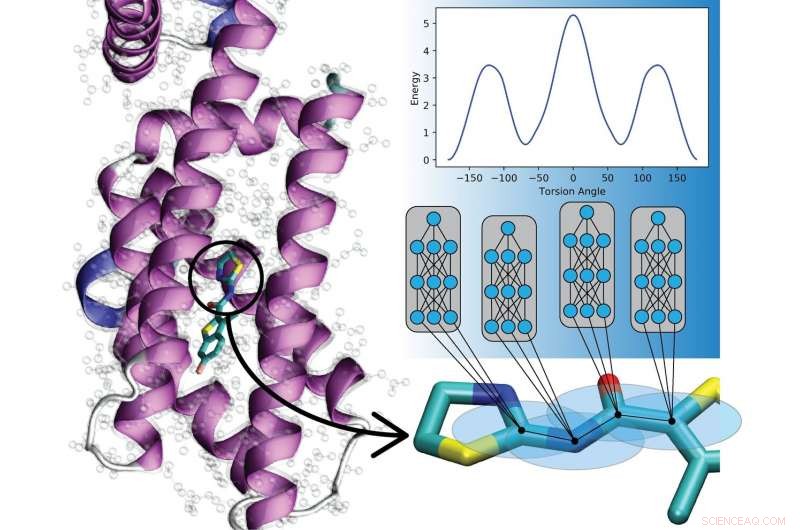
De nouveaux modèles d'apprentissage en profondeur prédisent les interactions entre les atomes dans les molécules organiques. Ces modèles aideront les biologistes computationnels et les chercheurs en développement de médicaments à comprendre et à traiter les maladies. Crédit :Laboratoire national de Los Alamos
Nouveau travail du Laboratoire national de Los Alamos, l'Université de Caroline du Nord à Chapel Hill, et l'Université de Floride montre que les réseaux neuronaux artificiels peuvent être entraînés à coder des lois de mécanique quantique pour décrire les mouvements des molécules, simulations de suralimentation potentiellement dans un large éventail de domaines.
"Cela signifie que nous pouvons désormais modéliser les matériaux et la dynamique moléculaire des milliards de fois plus rapidement que les méthodes quantiques conventionnelles, tout en conservant le même niveau de précision, " a déclaré Justin Smith, Physicien de Los Alamos et boursier Metropolis dans la division théorique du laboratoire. Comprendre comment les molécules se déplacent est essentiel pour exploiter leur valeur potentielle pour le développement de médicaments, simulations de protéines et chimie réactive, par exemple, et la mécanique quantique et les méthodes expérimentales (empiriques) alimentent les simulations.
La nouvelle technique, appelé potentiel ANI-1ccx, promet de faire progresser les capacités des chercheurs dans de nombreux domaines et d'améliorer la précision des potentiels basés sur l'apprentissage automatique dans les futures études sur les alliages métalliques et la physique des détonations.
Algorithmes de mécanique quantique (QM), utilisé sur les ordinateurs classiques, peut décrire avec précision les mouvements mécaniques d'un composé dans son environnement opérationnel. Mais QM évolue très mal avec des tailles moléculaires variables, limitant fortement le champ des simulations possibles. Même une légère augmentation de la taille moléculaire dans une simulation peut augmenter considérablement la charge de calcul. Ainsi, les praticiens recourent souvent à l'utilisation d'informations empiriques, qui décrit le mouvement des atomes en termes de physique classique et des lois de Newton, permettant des simulations à l'échelle de milliards d'atomes ou de millions de composés chimiques.
Traditionnellement, les potentiels empiriques ont dû trouver un compromis entre précision et transférabilité. Lorsque les nombreux paramètres du potentiel sont finement réglés pour un composé, la précision diminue sur d'autres composés.
Au lieu, l'équipe de Los Alamos, avec l'Université de Caroline du Nord à Chapel Hill et l'Université de Floride, a développé une approche d'apprentissage automatique appelée apprentissage par transfert qui leur permet de créer des potentiels empiriques en apprenant à partir de données collectées sur des millions d'autres composés. La nouvelle approche avec le potentiel empirique de l'apprentissage automatique peut être appliquée à de nouvelles molécules en quelques millisecondes, permettant la recherche d'un nombre beaucoup plus important de composés sur des échelles de temps beaucoup plus longues.