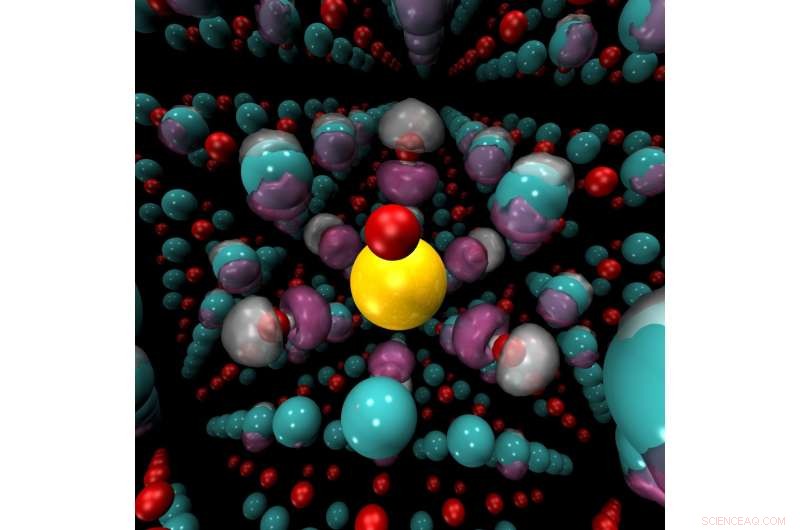
Contrainte mécanique, les changements de pression ou de température ou l'ajout d'agents dopants chimiques peuvent provoquer un passage brusque d'isolant à conducteur dans des matériaux tels que l'oxyde de nickel (illustré ici). Des ions nickel (bleu) et des ions oxygène (rouge) entourent un ion dopant potassium (jaune). Les méthodes de Monte Carlo quantique peuvent prédire avec précision les régions où la densité de charge (violet) s'accumulera dans ces matériaux. Crédit :Anouar Benali, Laboratoire National d'Argonne
Résoudre rapidement un problème complexe nécessite des compromis minutieux, et la simulation du comportement des matériaux ne fait pas exception. Pour obtenir des réponses qui prédisent le fonctionnement moléculaire de manière réalisable, les scientifiques doivent échanger des approximations mathématiques qui accélèrent les calculs au détriment de la précision.
Mais le magnétisme, la conductivité électrique et d'autres propriétés peuvent être assez délicates, dit Paul R.C. Kent du laboratoire national d'Oak Ridge du ministère de l'Énergie (DOE). Ces propriétés dépendent de la mécanique quantique, les mouvements et les interactions d'une myriade d'électrons et d'atomes qui forment les matériaux et déterminent leurs propriétés. Les chercheurs qui étudient de telles caractéristiques doivent modéliser de grands groupes d'atomes et de molécules plutôt que quelques-uns. La complexité de ce problème exige d'augmenter l'efficacité et la précision des outils de calcul.
C'est là qu'intervient une méthode appelée modélisation quantique de Monte Carlo (QMC). De nombreuses autres techniques se rapprochent du comportement des électrons en tant que moyenne globale, par exemple, plutôt que de les considérer individuellement. QMC permet de rendre compte du comportement individuel de tous les électrons sans approximations majeures, réduire les erreurs systématiques dans les simulations et produire des résultats fiables, dit Kent.
L'intérêt de Kent pour QMC remonte à son doctorat. recherche à l'Université de Cambridge dans les années 1990. A l'ORNL, il est récemment revenu à la méthode parce que les progrès du matériel des superordinateurs et des algorithmes avaient permis aux chercheurs d'améliorer sa précision.
"Nous pouvons créer de nouveaux matériaux et une fraction plus large d'éléments dans le tableau périodique, " dit Kent. " Plus important encore, nous pouvons commencer à faire certains des matériaux et des propriétés là où les méthodes plus approximatives que nous utilisons au jour le jour ne sont tout simplement pas fiables."
Même avec ces avancées, simulations de ces types de matériaux, ceux qui comprennent jusqu'à quelques centaines d'atomes et des milliers d'électrons, nécessite des calculs lourds. Kent dirige un centre des sciences de l'énergie de base du DOE, le Centre de Simulations Prédictives de Matériaux Fonctionnels (CPSFM) qui regroupe des chercheurs de l'ORNL, Laboratoire National d'Argonne, Laboratoires nationaux Sandia, Laboratoire national Lawrence Livermore, l'Université de Californie, Berkeley et Université d'État de Caroline du Nord.
Leur travail est soutenu par une allocation DOE Innovative and Novel Computational Impact on Theory and Experiments (INCITE) de 140 millions d'heures de processeur, répartis entre les superordinateurs Titan d'Oak Ridge Leadership Computing Facility et Mira d'Argonne Leadership Computing Facility. Les deux centres de calcul sont des installations pour les utilisateurs du DOE Office of Science.
Pour faire passer QMC au niveau supérieur, Kent et ses collègues commencent avec des matériaux tels que le dioxyde de vanadium qui présentent un comportement électronique inhabituel. À des températures plus fraîches, ce matériau isole contre le passage de l'électricité. Mais juste au-dessus de la température ambiante, le dioxyde de vanadium change brutalement sa structure et son comportement.
Soudain, ce matériau devient métallique et conduit efficacement l'électricité. Les scientifiques ne comprennent toujours pas exactement comment et pourquoi cela se produit. Des facteurs tels que les contraintes mécaniques, la pression ou le dopage des matériaux avec d'autres éléments induisent également cette transition rapide d'isolant à conducteur.
Cependant, si les scientifiques et les ingénieurs pouvaient contrôler ce comportement, ces matériaux pourraient être utilisés comme interrupteurs, capteurs ou, peut-être, la base des nouveaux appareils électroniques. "Ce grand changement de conductivité d'un matériau est le genre de chose que nous aimerions pouvoir prédire de manière fiable, " dit Kent.
Les chercheurs du laboratoire étudient également ces isolants-conducteurs avec des expériences. Cet effort de validation donne confiance à la puissance prédictive de leurs méthodes de calcul dans une gamme de matériaux. L'équipe a construit un logiciel open source, connu sous le nom de QMCPACK, qui est maintenant disponible en ligne et sur toutes les installations informatiques du DOE Office of Science.
Kent et ses collègues espèrent construire des supraconducteurs à haute température et d'autres matériaux complexes et mystérieux. Bien que les scientifiques connaissent les propriétés étendues de ces matériaux, Kent dit, "Nous ne pouvons pas encore les relier à la structure réelle et aux éléments des matériaux. C'est donc un très grand défi pour le domaine de la physique de la matière condensée."
Les méthodes de modélisation de la mécanique quantique les plus précises limitent les scientifiques à l'examen de quelques atomes ou molécules. Lorsque les scientifiques veulent étudier des systèmes plus grands, les coûts de calcul deviennent rapidement lourds. QMC propose un compromis :la taille d'un calcul augmente cubiquement par rapport au nombre d'électrons, un défi plus gérable. QMC n'intègre que quelques approximations contrôlées et peut être appliqué aux nombreux atomes et électrons nécessaires. Il est bien adapté aux supercalculateurs pétaflopiques d'aujourd'hui - capables d'effectuer au moins un milliard de calculs par seconde - et aux supercalculateurs exascale de demain, qui sera au moins mille fois plus rapide. La méthode mappe les éléments de simulation relativement facilement sur les nœuds de calcul dans ces systèmes.
L'équipe CPSFM continue d'optimiser QMCPACK pour des supercalculateurs toujours plus rapides, dont le Sommet de l'OLCF, qui sera pleinement opérationnel en janvier 2019. La capacité de mémoire plus élevée des GPU Nvidia Volta de cette machine – 16 gigaoctets par unité de traitement graphique contre 6 gigaoctets sur Titan – augmente déjà la vitesse de calcul. Avec l'aide d'Ed D'Azevedo et Andreas Tillack de l'OLCF, les chercheurs ont mis en œuvre des algorithmes améliorés qui peuvent doubler la vitesse de leurs calculs plus importants.
QMCPACK fait partie du projet d'informatique exascale du DOE, et l'équipe anticipe déjà des défis de mise à l'échelle supplémentaires pour l'exécution de QMCPACK sur les futures machines. Pour effectuer les simulations souhaitées en 12 heures environ sur un supercalculateur exascale, Kent estime qu'ils auront besoin d'algorithmes 30 fois plus évolutifs que ceux de la version actuelle.
Même avec du matériel et des algorithmes améliorés, Les calculs QMC seront toujours coûteux. Kent et son équipe aimeraient donc utiliser QMCPACK pour comprendre où les méthodes moins chères ne fonctionnent pas afin de pouvoir les améliorer. Ensuite, ils peuvent enregistrer les calculs QMC pour les problèmes les plus difficiles en science des matériaux, dit Kent. « Idéalement, nous apprendrons ce qui rend ces matériaux très difficiles à modéliser, puis améliorerons les approches moins chères afin que nous puissions faire des analyses beaucoup plus larges de différents matériaux. »
La combinaison de méthodes QMC améliorées et d'une suite d'approches de modélisation moins coûteuses en calcul pourrait ouvrir la voie à de nouveaux matériaux et à une compréhension de leurs propriétés. Concevoir et tester de nouveaux composés en laboratoire coûte cher, dit Kent. Les scientifiques pourraient économiser un temps et des ressources précieux s'ils pouvaient d'abord prédire le comportement de nouveaux matériaux dans une simulation.
Plus, note-t-il, des méthodes de calcul fiables pourraient aider les scientifiques à comprendre les propriétés et les processus qui dépendent d'atomes individuels qui sont extrêmement difficiles à observer à l'aide d'expériences. "C'est un endroit où il y a beaucoup d'intérêt à aller après la science fondamentale, prédire de nouveaux matériaux et permettre des applications technologiques."