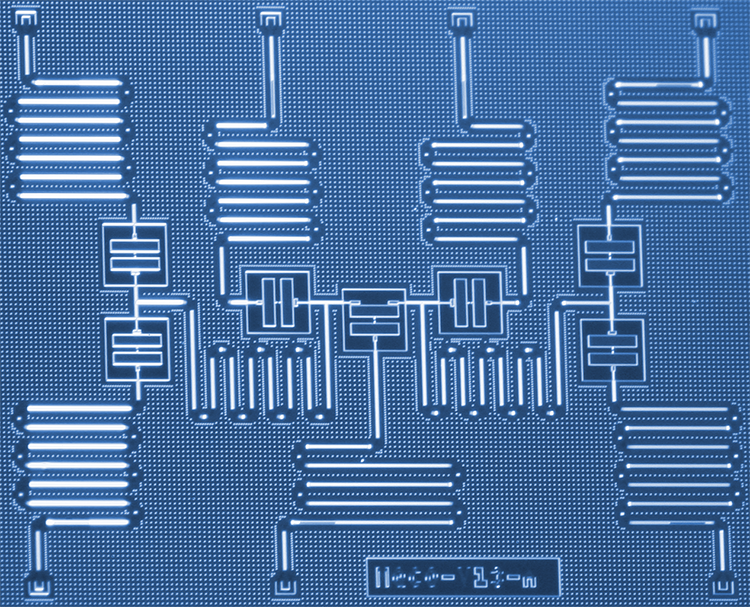
Les scientifiques d'IBM ont développé une nouvelle approche pour simuler des molécules sur un ordinateur quantique qui pourrait un jour révolutionner la chimie et la science des matériaux. Les scientifiques ont utilisé avec succès six qubits sur un processeur quantique à sept qubits spécialement conçu pour résoudre le problème de structure moléculaire de l'hydrure de béryllium (BeH2) - la plus grande molécule simulée sur un ordinateur quantique à ce jour. Les résultats démontrent une voie d'exploration pour les systèmes quantiques à court terme afin d'améliorer notre compréhension des réactions chimiques complexes qui pourraient conduire à des applications pratiques. Crédit :Kandala et al.; La nature
La simulation de molécules sur des ordinateurs quantiques est devenue beaucoup plus facile avec le matériel quantique supraconducteur d'IBM. Dans un récent article de recherche publié dans La nature , Solveur propre quantique variationnel à efficacité matérielle pour les petites molécules et les aimants quantiques, nous implémentons un nouvel algorithme quantique capable de calculer efficacement l'état d'énergie le plus bas des petites molécules. En cartographiant la structure électronique des orbitales moléculaires sur un sous-ensemble de notre processeur quantique à sept qubits spécialement conçu, nous avons étudié des molécules jusqu'alors inexplorées par les ordinateurs quantiques, y compris l'hydrure de lithium (LiH) et l'hydrure de béryllium (BeH2). L'encodage particulier des orbitales aux qubits étudié dans ce travail peut être utilisé pour simplifier les simulations de molécules encore plus grosses et nous espérons avoir l'opportunité d'explorer de telles simulations plus grandes à l'avenir, lorsque la puissance de calcul quantique (ou « volume quantique ») des systèmes IBM Q a augmenté.
Alors que BeH2 est la plus grosse molécule jamais simulée par un ordinateur quantique à ce jour, le modèle considéré de la molécule elle-même est encore assez simple pour que les ordinateurs classiques puissent simuler exactement. Cela en a fait un cas de test pour repousser les limites de ce que notre processeur à sept qubits pouvait réaliser, approfondir notre compréhension des exigences pour améliorer la précision de nos simulations quantiques, et poser les éléments fondamentaux nécessaires à l'exploration de telles études sur l'énergie moléculaire.
Les meilleures simulations de molécules aujourd'hui sont exécutées sur des ordinateurs classiques qui utilisent des méthodes approximatives complexes pour estimer l'énergie la plus basse d'un hamiltonien moléculaire. Un "Hamiltonien" est un opérateur d'énergie mécanique quantique qui décrit les interactions entre toutes les orbitales électroniques et les noyaux des atomes constitutifs. L'état "d'énergie la plus basse" de l'hamiltonien moléculaire dicte la structure de la molécule et la façon dont elle interagira avec d'autres molécules. Ces informations sont essentielles pour que les chimistes conçoivent de nouvelles molécules, réactions, et les procédés chimiques pour les applications industrielles.
Qubit :Orbital
Bien que notre processeur quantique à sept qubits ne soit pas entièrement à correction d'erreurs et tolérant aux pannes, les temps de cohérence des qubits individuels durent environ 50 µs. Il est donc très important d'utiliser un algorithme quantique très efficace pour tirer le meilleur parti de notre précieuse cohérence quantique et essayer de comprendre les structures moléculaires. L'algorithme doit être efficace en termes de nombre de qubits utilisés et de nombre d'opérations quantiques effectuées.

Application à la chimie quantique. a-c, Résultats expérimentaux (cercles noirs remplis), surfaces d'énergie exactes (lignes pointillées) et tracés de densité (ombrage ; voir échelles de couleurs) des résultats des simulations numériques, pour plusieurs distances interatomiques pour H2 (a), LiH (b) et BeH2 (c). Les résultats expérimentaux et numériques présentés sont pour des circuits de profondeur d = 1 Les barres d'erreur sur les données expérimentales sont plus petites que la taille des marqueurs. Les tracés de densité sont obtenus à partir de 100 résultats numériques à chaque distance interatomique. Les encarts supérieurs de chaque panneau mettent en évidence les qubits utilisés pour l'expérience et les portes de résonance croisée (flèches, étiqueté CRc–t; où « c » désigne le qubit de contrôle et « t » le qubit cible) qui constituent UENT. Les encarts inférieurs sont des représentations de la géométrie moléculaire (pas à l'échelle). Pour les trois molécules, l'écart des résultats expérimentaux par rapport aux courbes exactes est bien expliqué par les simulations stochastiques. Crédit :Kandala et al.; La nature
Notre schéma contraste avec les algorithmes de simulation quantique précédemment étudiés, qui se concentrent sur l'adaptation des schémas de simulation moléculaire classiques au matériel quantique - et ce faisant, ne prennent pas efficacement en compte les frais généraux limités des dispositifs quantiques réalistes actuels.
Ainsi, au lieu de forcer les méthodes de calcul classiques sur du matériel quantique, nous avons inversé l'approche et demandé :comment pouvons-nous extraire la puissance de calcul quantique maximale de notre processeur à sept qubits ?
Notre réponse à cela combine un certain nombre de techniques efficaces en termes de matériel pour attaquer le problème :
Avec les futurs processeurs quantiques, qui aura plus de volume quantique, nous pourrons explorer la puissance de cette approche de la simulation quantique pour des molécules de plus en plus complexes qui dépassent les capacités de calcul classiques. La capacité de simuler avec précision des réactions chimiques, est propice aux efforts de découverte de nouveaux médicaments, les engrais, même de nouvelles sources d'énergie durables.
Les expériences que nous détaillons dans notre article n'ont pas été exécutées sur nos processeurs actuellement disponibles publiquement à cinq qubits et à 16 qubits sur le cloud. Mais les développeurs et les utilisateurs de l'expérience IBM Q peuvent désormais accéder aux cahiers Jupyter de chimie quantique sur le référentiel github QISKit. Sur le système à cinq qubits, les utilisateurs peuvent explorer la simulation d'énergie à l'état fondamental pour les petites molécules d'hydrogène et de LiH. Des cahiers pour les molécules plus grosses sont disponibles pour ceux qui ont un accès bêta au processeur 16 qubit mis à niveau.