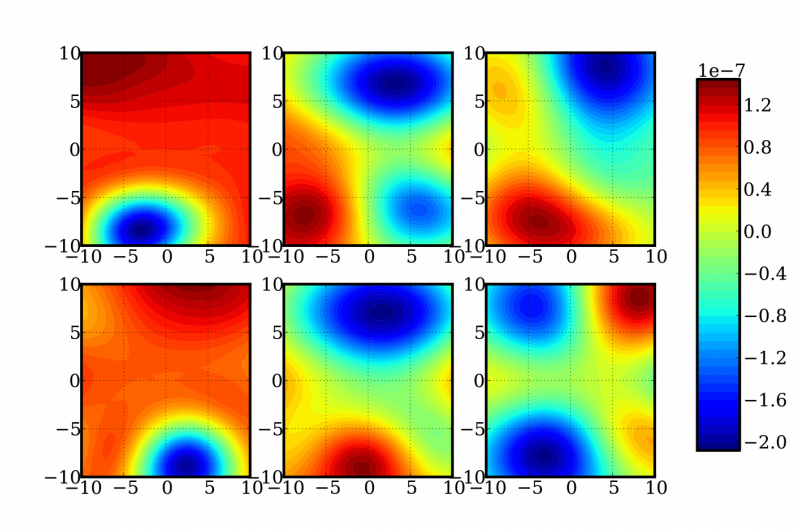
Une représentation de tranches bidimensionnelles aléatoires d'une fonction à 12 dimensions pour déterminer les corrections d'énergie et de fréquence d'une molécule de formaldéhyde. Crédit :Laboratoires nationaux Sandia
Des chercheurs des laboratoires nationaux Sandia ont développé de nouvelles techniques mathématiques pour faire progresser l'étude des molécules au niveau quantique.
Des développements mathématiques et algorithmiques dans ce sens sont nécessaires pour permettre l'étude détaillée des molécules d'hydrocarbures complexes qui sont pertinentes dans la combustion des moteurs.
Les méthodes existantes pour approcher les fonctions d'énergie potentielle à l'échelle quantique nécessitent trop de puissance informatique et sont donc limitées aux petites molécules. Les chercheurs de Sandia affirment que leur technique accélérera les calculs de mécanique quantique et améliorera les prédictions faites par les modèles de chimie théorique. Compte tenu de l'accélération de calcul, ces méthodes peuvent potentiellement être appliquées à des molécules plus grosses.
Le chercheur postdoctoral de Sandia, Prashant Rai, a travaillé avec les chercheurs Khachik Sargsyan et Habib Najm du centre de recherche sur la combustion de Sandia et a collaboré avec les chimistes quantiques So Hirata et Matthew Hermes de l'Université de l'Illinois à Urbana-Champaign. Calculer l'énergie à moins d'arrangements géométriques que normalement requis, l'équipe a développé des méthodes informatiques efficaces pour approcher les surfaces d'énergie potentielle.
Une compréhension précise des surfaces énergétiques potentielles, éléments clés dans pratiquement tous les calculs de la dynamique quantique, est nécessaire pour estimer avec précision l'énergie et la fréquence des modes de vibration des molécules.
"Si nous pouvons trouver l'énergie de la molécule pour toutes les configurations possibles, nous pouvons déterminer des informations importantes, tels que les états stables de la structure de transition moléculaire ou les états intermédiaires des molécules dans les réactions chimiques, " dit Raï.
Les premiers résultats de cette recherche ont été publiés dans Physique Moléculaire dans un article intitulé « Décomposition en tenseur canonique de bas rang des surfaces d'énergie potentielle :application à la théorie de la fonction de Green vibrationnelle schématique basée sur la grille ».

Prashant Rai, chercheurs des Laboratoires nationaux Sandia, la gauche, Habib Najm, centre, et Khachik Sargsyan discutent des techniques mathématiques utilisées pour étudier le comportement de grosses molécules à l'échelle quantique. Crédit :Dino Vournas
" L'approximation des surfaces d'énergie potentielle de molécules plus grosses est une tâche extrêmement difficile en raison de l'augmentation exponentielle des informations nécessaires pour les décrire avec chaque atome supplémentaire dans le système, " dit Rai. " En mathématiques, c'est ce qu'on appelle la malédiction de la dimensionnalité."
Battre la malédiction
La clé pour vaincre la malédiction de la dimensionnalité est d'exploiter les caractéristiques de la structure spécifique des surfaces d'énergie potentielle. Rai a déclaré que ces informations de structure peuvent ensuite être utilisées pour approximer les fonctions de haute dimension requises.
"Nous utilisons le fait que bien que les surfaces d'énergie potentielle puissent être de grande dimension, ils peuvent être bien approchés comme une petite somme de produits de fonctions unidimensionnelles. C'est ce qu'on appelle la structure de bas rang, où le rang de la surface d'énergie potentielle est le nombre de termes de la somme, " Rai a dit. " Une telle hypothèse sur la structure est assez générale et a également été utilisée dans des problèmes similaires dans d'autres domaines. Mathématiquement, l'intuition des techniques d'approximation de bas rang vient de l'algèbre multilinéaire où la fonction est interprétée comme un tenseur et est décomposée à l'aide de techniques de décomposition tensorielles standard."
Les corrections d'énergie et de fréquence sont formulées comme des intégrales de ces fonctions énergétiques de grande dimension. L'approximation dans un format de rang aussi bas rend ces fonctions facilement intégrables car elle brise le problème d'intégration à la somme des produits d'intégrales à une ou deux dimensions, les méthodes d'intégration standard s'appliquent donc.
L'équipe a testé ses méthodes de calcul sur de petites molécules telles que l'eau et le formaldéhyde. Par rapport à la méthode Monte Carlo classique, le cheval de bataille standard basé sur l'aléatoire pour les problèmes d'intégration de grande dimension, leur approche a prédit l'énergie et la fréquence de la molécule d'eau qui étaient plus précises, et c'était au moins 1, 000 fois plus efficace en calcul.
Rai a déclaré que la prochaine étape consiste à améliorer encore la technique en la défiant avec des molécules plus grosses, comme le benzène.
"Études interdisciplinaires, comme la chimie quantique et le génie de la combustion, offrir des opportunités de pollinisation croisée des idées, offrant ainsi une nouvelle perspective sur les problèmes et leurs solutions possibles, ", a déclaré Rai. "C'est également une étape vers l'utilisation des progrès récents de la science des données comme pilier de la découverte scientifique à l'avenir."