L'équipe de Jianwei Shuai et l'équipe de Jiahuai Han de l'Université de Xiamen ont développé un logiciel d'analyse de données d'acquisition indépendant des données, basé sur un auto-encodeur profond, pour la spectrométrie de masse des protéines, qui réalise l'analyse de peptides et de protéines pertinents à partir de données de spectrométrie de masse de protéines complexes, et démontre la supériorité et polyvalence de la méthode sur différents instruments et échantillons d’espèces. L'étude a été publiée dans Research comme "Cher-DIA XMBD :auto-encodeur profond pour la protéomique d'acquisition indépendante des données".
Les protéines jouent un rôle central en tant qu’exécutrices des activités de la vie cellulaire, pilotant une myriade de processus biologiques cruciaux. Par conséquent, le domaine de la protéomique a fait l’objet d’une large attention. La protéomique implique l'étude approfondie des propriétés des protéines, y compris les modifications post-traductionnelles, les niveaux d'expression des protéines, les interactions protéine-protéine, etc. Son objectif global est d'acquérir une compréhension holistique de la pathogenèse des maladies, du métabolisme cellulaire et d'autres processus vitaux au niveau protéique.
Parmi les techniques analytiques clés de la recherche en protéomique, la spectrométrie de masse des protéines se démarque comme la plus critique. Au fil du temps, la technologie de spectrométrie de masse a évolué pour fournir aux chercheurs des outils fiables et dynamiques pour l'analyse protéomique.
Deux approches principales de la spectrométrie de masse des protéines sont l'acquisition dépendante des données (DDA) et l'acquisition indépendante des données (DIA). Dans DDA, tous les spectres d'ions précurseurs peptidiques (MS1) sont acquis en mode d'analyse complète, suivi de la sélection des ions peptidiques les plus intensifs en N pour la fragmentation afin d'obtenir des spectres d'ions fragments (MS2).
Malgré son utilité, le DDA est confronté à des défis liés à la reproductibilité expérimentale et à la détection de peptides de faible abondance en raison du caractère aléatoire de la fragmentation peptidique et de la sélection préférentielle de peptides de haute intensité.
Pour surmonter ces limitations, la méthode d'acquisition DIA a été introduite. Cette technique divise la plage du rapport masse/charge des spectres d’ions parents en plusieurs fenêtres et fragmente séquentiellement tous les peptides dans chaque fenêtre pour obtenir des spectres d’ions filles. Une méthode DIA courante est l'acquisition séquentielle par fenêtre de tous les ions fragments théoriques (SWATH).
Bien que les données d'acquisition DIA conservent des informations protéomiques plus complètes, leur grande taille de données, leur haute dimensionnalité et leurs signaux spectraux complexes posent des défis à leur analyse. En conséquence, l'exploration de données DIA est devenue un objectif majeur dans la communauté protéomique.
L'équipe de Jianwei Shuai et celle de Jiahuai Han ont collaboré pour développer Dear-DIA, un logiciel d'analyse de données d'acquisition indépendant des données basé sur l'apprentissage profond, qui réalise l'identification d'ions fragments correspondant à différents peptides à partir de spectres d'acquisition DIA complexes et démontre la généralisation à des échantillons complexes de différentes espèces.
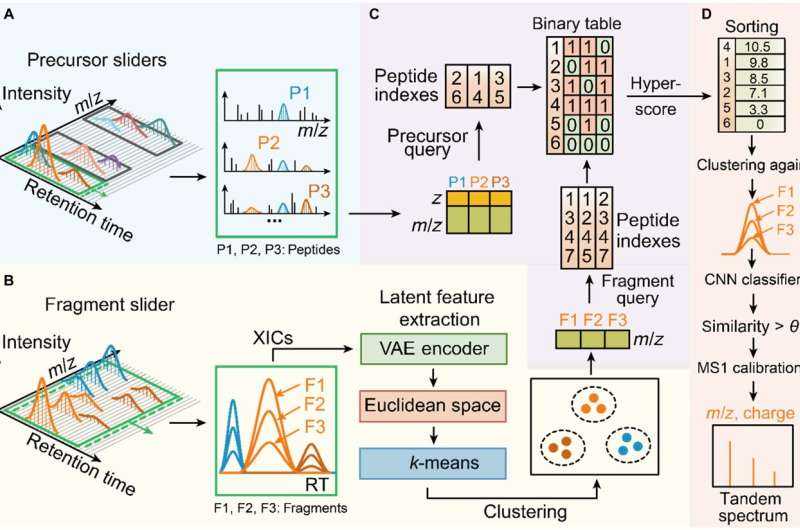
Dear-DIA divise d'abord les spectres en un curseur de largeur fixe avec une largeur fixe dans la direction du temps de rétention (RT), et chaque curseur contient un ensemble de spectres de précurseurs MS1 et de spectres de fragments MS2 comme unité de traitement minimale. Ensuite, un algorithme de recherche de pic a été utilisé pour éliminer les ions de fond à faible rapport signal/bruit et conserver les ions précurseurs candidats et les ions fragments candidats.
Ensuite, Dear-DIA utilise un auto-encodeur variationnel pour extraire les caractéristiques maximales des ions fragments et cartographie les caractéristiques dans l'espace euclidien, puis regroupe les caractéristiques, avec différentes classes de fragments correspondant à différents peptides, réalisant ainsi le processus de déconvolution du spectrogramme.
Dear-DIA comprend un algorithme d'indexation appelé PIndex, qui fait correspondre les précurseurs aux résultats de regroupement de fragments et sélectionne les meilleurs résultats d'appariement par notation. Dear-DIA utilise un réseau neuronal convolutif pour recalculer la similarité de forme des pics des fragments de la même classe afin d'éliminer les ions interférents et de regrouper les résultats avec une faible similarité.
Les auteurs ont d'abord testé les performances de Dear-DIA sur un ensemble de données SGS Human contenant 422 peptides synthétiques d'étalons marqués par des isotopes stables, divisés en 10 gradients de dilution (de 1 à 512 fois), et les données DIA ont été obtenues sur un AB. Spectromètre de masse SCIEX TTOF5600 utilisant la technique SWATH pour obtenir des données DIA.
Les résultats de l'analyse ont montré que Dear-DIA a trouvé plus de peptides synthétiques dans toutes les solutions diluées par rapport aux deux méthodes analytiques couramment utilisées, Spectronaut 14 et DIA-Umpire. Les auteurs ont également comparé le nombre de peptides et de protéines trouvés par les différentes méthodes analytiques pour les ensembles de données SGS Human et L929 Mouse. Les résultats ont montré que Dear-DIA était capable de trouver plus de peptides et de protéines que Spectronaut 14 et DIA-Umpire, couvrant plus de 85 % de leurs résultats.
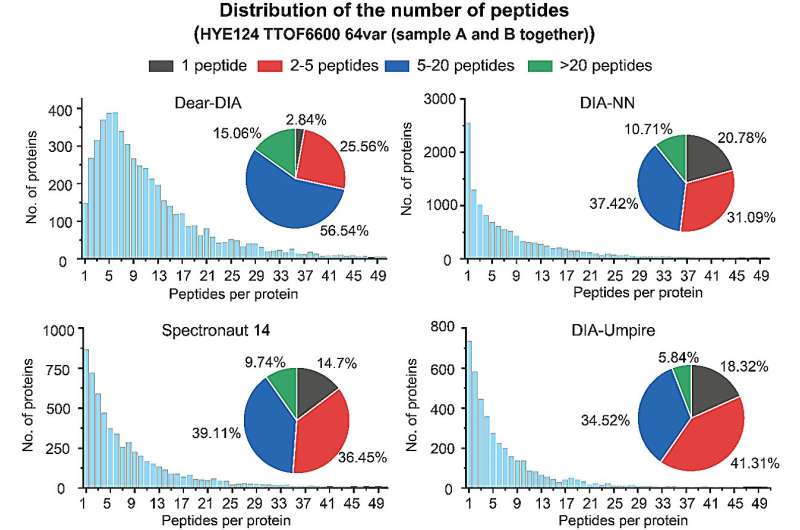
La fiabilité des résultats de l’analyse protéomique peut également être démontrée par le nombre de peptides identifiés pour chaque protéine. Les protéines avec 2 peptides identifiés ou plus sont généralement considérées comme des identifications plus crédibles. Les auteurs ont comparé le nombre de protéines par rapport aux peptides rapportés par Dear-DIA avec un logiciel existant sur un ensemble de données d'espèces mixtes (ensemble de données HYE124 TTOF6600 64var).
L'ensemble de données contient des protéines de trois espèces, l'humain, la levure et E. coli, et les données ont été acquises sur un spectromètre de masse AB SCIEX TTOF6600 en utilisant la méthode SWATH, avec des spectres d'ions parents contenant 64 fenêtres variables. Les résultats de l'analyse ont montré que 97,16 % des protéines trouvées par Dear-DIA pourraient correspondre à 2 peptides ou plus, ce qui est beaucoup plus élevé que DIA-NN, Spectronaut 14 et DIA-Umpire.
Les techniques d’acquisition indépendantes des données pour la protéomique ont été largement adoptées et les algorithmes d’analyse associés sont devenus un point chaud de la recherche. La découverte de protéines à partir de données massives de spectrométrie de masse est une tâche intéressante et stimulante. Dans cet article, l'équipe a développé Dear-DIA, un logiciel d'analyse basé sur l'apprentissage profond, utilisé pour traiter une variété de données d'acquisition DIA très complexes et capable de découvrir davantage de peptides et de protéines, en plus de reproduire la plupart des résultats de Spectronaut et DIA-Arbitre.
De plus, bien que l'ensemble de données de formation provienne d'E. coli, les excellentes performances de Dear-DIA sur l'ensemble de données d'espèces mixtes démontrent sa forte capacité de généralisation à analyser des données protéomiques complexes. L'apprentissage profond, en tant qu'outil largement utilisé pour l'analyse du Big Data, a démontré d'excellentes capacités d'exploration de données pour découvrir des associations intrinsèques profondes dans le Big Data.
L'utilisation de l'apprentissage profond pour analyser les données de spectrométrie de masse protéomique présente un grand potentiel et favorisera davantage l'étude de questions fondamentales telles que les réseaux de signalisation des protéines.
Plus d'informations : Qingzu He et al, Dear-DIA XMBD :Un encodeur automatique profond permet la déconvolution de la protéomique d'acquisition indépendante des données, Recherche (2023). DOI :10.34133/research.0179
Informations sur le journal : Recherche
Fourni par Recherche