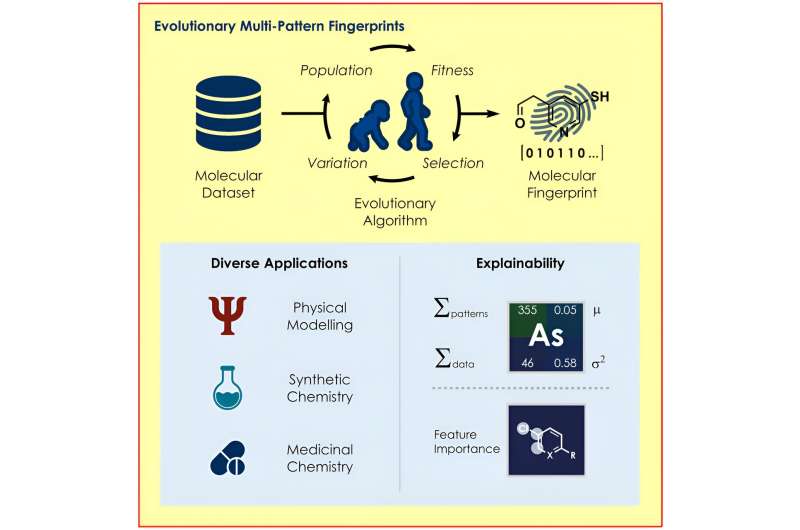
Une équipe dirigée par le professeur Frank Glorius de l'Institut de chimie organique de l'Université de Münster a développé un algorithme évolutif qui identifie les structures d'une molécule particulièrement pertinentes pour une question donnée et les utilise pour coder les propriétés des molécules pour diverses questions. modèles d'apprentissage automatique.
La méthode convient également à la prédiction automatique des propriétés chimiques quantiques et de la toxicité des molécules. Il peut être appliqué à n'importe quel ensemble de données moléculaires et ne nécessite aucune connaissance experte des relations sous-jacentes.
L’intelligence artificielle et l’apprentissage automatique deviennent de plus en plus pertinents dans la vie quotidienne, et il en va de même pour la chimie. Les chimistes organiques, par exemple, s'intéressent à la manière dont l'apprentissage automatique peut aider à découvrir et à synthétiser de nouvelles molécules efficaces contre les maladies ou utiles à d'autres égards.
Le nouvel algorithme développé par l'équipe de Glorius recherche des représentations moléculaires optimales basées sur les principes de l'évolution, en utilisant des mécanismes tels que la reproduction, la mutation et la sélection. En fonction du modèle et de la question posée, des « empreintes moléculaires » personnalisées sont créées, que les chimistes ont utilisées dans leur étude pour prédire les réactions chimiques avec une précision surprenante.
La méthode, publiée dans la revue Chem , convient également pour prédire les propriétés chimiques quantiques et la toxicité des molécules.
Afin d’utiliser l’apprentissage automatique, les chercheurs doivent d’abord convertir les molécules sous une forme lisible par ordinateur. De nombreux groupes de recherche se sont déjà attaqués à ce problème et, par conséquent, il existe différentes manières de réaliser cette tâche. Cependant, il est difficile de prédire laquelle des méthodes disponibles est la mieux adaptée pour répondre à une question spécifique, par exemple pour déterminer si un composé chimique est nocif pour l'homme.
Le nouvel algorithme est conçu pour aider à trouver l’empreinte moléculaire optimale dans chaque cas. Pour ce faire, l'algorithme sélectionne progressivement les empreintes moléculaires qui obtiennent les meilleurs résultats de prédiction parmi de nombreuses empreintes moléculaires générées aléatoirement.
"À l'instar de la nature, nous utilisons des mutations, c'est-à-dire des modifications aléatoires de composants individuels des empreintes digitales, ou recombinons des composants de deux empreintes digitales", explique le doctorant Felix Katzenburg.
"Dans d'autres études, les molécules sont souvent décrites par des propriétés quantifiables qui ont été sélectionnées et calculées par des humains", ajoute Glorius.
"Étant donné que l'algorithme que nous avons développé identifie automatiquement les structures moléculaires pertinentes, il n'y a aucun biais systématique causé par des experts humains."
Un autre avantage est que la méthode de codage permet de comprendre pourquoi un modèle fait une certaine prédiction. Par exemple, il est possible de tirer des conclusions sur les parties d'une molécule qui ont un impact positif ou négatif sur la prédiction du déroulement d'une réaction, permettant ainsi aux chercheurs de modifier les structures pertinentes de manière ciblée.
L'équipe de Münster a constaté que sa nouvelle méthode n'obtenait pas toujours les résultats les plus optimaux.
"Lorsqu'une expertise humaine considérable a été consacrée à la sélection de propriétés moléculaires particulièrement pertinentes ou que de très grandes quantités de données sont disponibles, d'autres méthodes telles que les réseaux neuronaux ont parfois l'avantage", explique Katzenburg.
Cependant, l'un des principaux objectifs de l'étude était de développer une méthode de codage de molécules pouvant être appliquée à n'importe quel ensemble de données moléculaires et ne nécessitant pas de connaissances spécialisées sur les relations sous-jacentes.
Plus d'informations : Philipp M. Pflüger et al, Un algorithme évolutif pour des représentations moléculaires interprétables, Chem (2024). DOI :10.1016/j.chempr.2024.02.004
Informations sur le journal : Chimie
Fourni par l'Université de Münster