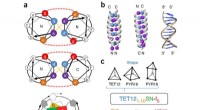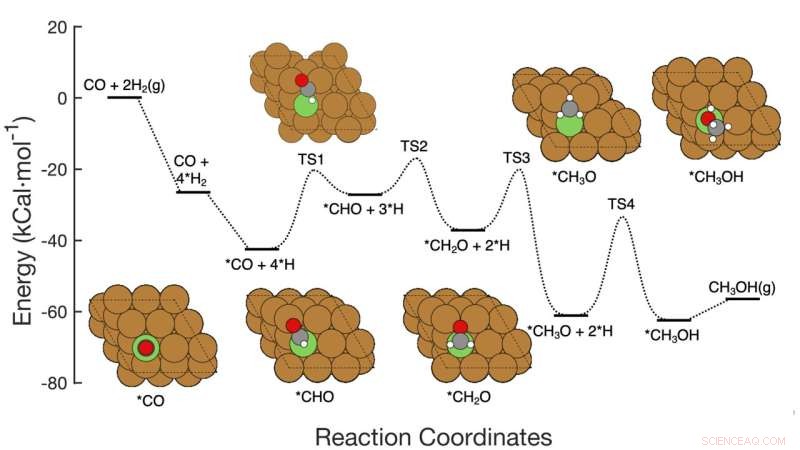
Ce graphique montre la voie de réaction en sept étapes de l'hydrogénation du CO en méthanol sur des catalyseurs à base de cuivre, y compris les réactifs à chaque étape, les arrangements atomiques schématiques des intermédiaires et les barrières d'activation énergétique nécessaires pour passer d'une étape à l'autre. L'équipe du Brookhaven Lab a présenté un cadre d'apprentissage automatique qui a identifié avec succès les étapes/combinaisons d'étapes à modifier pour améliorer la production de méthanol. Leur travail pourrait aider à guider la conception de nouveaux catalyseurs pour atteindre cet objectif et le cadre peut être appliqué pour optimiser d'autres réactions. Crédit :Laboratoire national de Brookhaven
Les chimistes du laboratoire national de Brookhaven du département américain de l'énergie ont développé un nouveau cadre d'apprentissage automatique (ML) qui peut se concentrer sur les étapes d'une conversion chimique en plusieurs étapes qui doivent être modifiées pour améliorer la productivité. L'approche pourrait aider à guider la conception de catalyseurs - des « négociateurs » chimiques qui accélèrent les réactions.
L'équipe a développé la méthode pour analyser la conversion du monoxyde de carbone (CO) en méthanol à l'aide d'un catalyseur à base de cuivre. La réaction consiste en sept étapes élémentaires assez simples.
"Notre objectif était d'identifier quelle étape élémentaire du réseau de réaction ou quel sous-ensemble d'étapes contrôle l'activité catalytique", a déclaré Wenjie Liao, le premier auteur d'un article décrivant la méthode qui vient d'être publiée dans la revue Catalysis Science &Technology . Liao est un étudiant diplômé de l'Université de Stony Brook qui a travaillé avec des scientifiques du groupe Catalysis Reactivity and Structure (CRS) de la division de chimie du Brookhaven Lab.
Ping Liu, le chimiste du CRS qui a dirigé les travaux, a déclaré :"Nous avons utilisé cette réaction comme exemple de notre méthode de cadre ML, mais vous pouvez mettre n'importe quelle réaction dans ce cadre en général."
Cibler les énergies d'activation
Imaginez une réaction chimique en plusieurs étapes comme des montagnes russes avec des collines de différentes hauteurs. La hauteur de chaque colline représente l'énergie nécessaire pour passer d'un pas à l'autre. Les catalyseurs abaissent ces "barrières d'activation" en facilitant la rencontre des réactifs ou en leur permettant de le faire à des températures ou des pressions plus basses. Pour accélérer la réaction globale, un catalyseur doit cibler l'étape ou les étapes qui ont le plus grand impact.
Traditionnellement, les scientifiques cherchant à améliorer une telle réaction calculaient comment changer chaque barrière d'activation une à la fois pourrait affecter le taux de production global. Ce type d'analyse pourrait identifier quelle étape était "limitante" et quelles étapes déterminent la sélectivité de la réaction, c'est-à-dire si les réactifs se dirigent vers le produit souhaité ou vers une voie alternative vers un sous-produit indésirable.

Ping Liu, chimiste du Brookhaven Lab, et Wenjie Liao, étudiante diplômée à l'Université de Stony Brook, ont développé un cadre d'apprentissage automatique pour identifier les étapes de réaction chimique qui pourraient être ciblées pour améliorer la productivité de la réaction. Crédit :Laboratoire national de Brookhaven
Mais, selon Liu, "Ces estimations finissent par être très approximatives avec beaucoup d'erreurs pour certains groupes de catalyseurs. Cela a vraiment nui à la conception et au criblage des catalyseurs, ce que nous essayons de faire", a-t-elle déclaré.
Le nouveau cadre d'apprentissage automatique est conçu pour améliorer ces estimations afin que les scientifiques puissent mieux prédire comment les catalyseurs affecteront les mécanismes de réaction et la production chimique.
"Maintenant, au lieu de déplacer une barrière à la fois, nous déplaçons toutes les barrières simultanément. Et nous utilisons l'apprentissage automatique pour interpréter cet ensemble de données", a déclaré Liao.
Cette approche, selon l'équipe, donne des résultats beaucoup plus fiables, y compris sur la façon dont les étapes d'une réaction fonctionnent ensemble.
« Dans des conditions de réaction, ces étapes ne sont pas isolées ou séparées les unes des autres; elles sont toutes connectées », a déclaré Liu. "Si vous ne faites qu'une étape à la fois, vous manquez beaucoup d'informations :les interactions entre les étapes élémentaires. C'est ce qui a été capturé dans ce développement", a-t-elle déclaré.
Construire le modèle
Les scientifiques ont commencé par créer un ensemble de données pour former leur modèle d'apprentissage automatique. L'ensemble de données était basé sur les calculs de la "théorie fonctionnelle de la densité" (DFT) de l'énergie d'activation nécessaire pour transformer un arrangement d'atomes en un autre à travers les sept étapes de la réaction. Ensuite, les scientifiques ont effectué des simulations sur ordinateur pour explorer ce qui se passerait s'ils modifiaient simultanément les sept barrières d'activation :certaines montaient, d'autres descendaient, certaines individuellement et d'autres par paires.
"La gamme de données que nous avons incluse était basée sur l'expérience antérieure avec ces réactions et ce système catalytique, dans la gamme intéressante de variation qui est susceptible de vous donner de meilleures performances", a déclaré Liu.
En simulant des variations dans 28 "descripteurs" - y compris les énergies d'activation pour les sept étapes plus des paires d'étapes changeant deux à la fois - l'équipe a produit un ensemble de données complet de 500 points de données. Cet ensemble de données a prédit comment tous ces ajustements individuels et ces paires d'ajustements affecteraient la production de méthanol. Le modèle a ensuite noté les 28 descripteurs en fonction de leur importance dans la production de méthanol.
"Notre modèle a " appris " des données et identifié six descripteurs clés qui, selon lui, auraient le plus d'impact sur la production ", a déclaré Liao.
Une fois les descripteurs importants identifiés, les scientifiques ont recyclé le modèle ML en utilisant uniquement ces six descripteurs "actifs". Ce modèle ML amélioré a pu prédire l'activité catalytique en se basant uniquement sur des calculs DFT pour ces six paramètres.
« Plutôt que de devoir calculer l'ensemble des 28 descripteurs, vous pouvez désormais calculer avec seulement les six descripteurs et obtenir les taux de conversion du méthanol qui vous intéressent », a déclaré Liu.
L'équipe dit qu'elle peut également utiliser le modèle pour cribler les catalyseurs. S'ils peuvent concevoir un catalyseur qui améliore la valeur des six descripteurs actifs, le modèle prédit un taux de production de méthanol maximal.
Comprendre les mécanismes
Lorsque l'équipe a comparé les prédictions de leur modèle avec les performances expérimentales de leur catalyseur - et les performances des alliages de divers métaux avec du cuivre - les prédictions correspondaient aux résultats expérimentaux. Les comparaisons de l'approche ML avec la méthode précédente utilisée pour prédire les performances des alliages ont montré que la méthode ML était bien supérieure.
Les données ont également révélé de nombreux détails sur la manière dont les changements dans les barrières énergétiques pourraient affecter le mécanisme de réaction. La manière dont les différentes étapes de la réaction fonctionnent ensemble est particulièrement intéressante et importante. Par exemple, les données ont montré que, dans certains cas, l'abaissement de la barrière énergétique dans l'étape de limitation de vitesse seule n'améliorerait pas en soi la production de méthanol. Mais modifier la barrière énergétique d'une étape plus tôt dans le réseau de réaction, tout en maintenant l'énergie d'activation de l'étape limitant la vitesse dans une plage idéale, augmenterait la production de méthanol.
"Notre méthode nous donne des informations détaillées que nous pourrions utiliser pour concevoir un catalyseur qui coordonne bien l'interaction entre ces deux étapes", a déclaré Liu.
Mais Liu est très enthousiasmé par la possibilité d'appliquer ces cadres de ML basés sur les données à des réactions plus complexes.
"Nous avons utilisé la réaction au méthanol pour démontrer notre méthode. Mais la façon dont elle génère la base de données et comment nous formons le modèle ML et comment nous interpolons le rôle de la fonction de chaque descripteur pour déterminer le poids global en termes d'importance, cela peut être s'applique facilement à d'autres réactions », a-t-elle déclaré. Découverte d'un nouveau catalyseur pour l'hydrogénation hautement active et sélective du dioxyde de carbone en méthanol