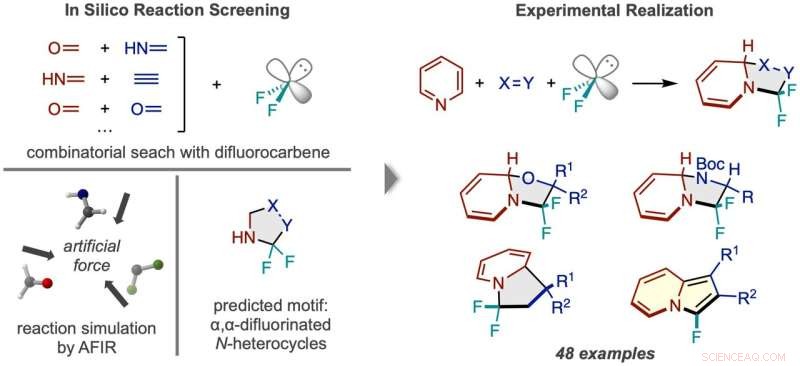
Workflow de découverte de réactions via un criblage in silico. (À gauche) Des réactions entre le difluorocarbène et de nombreuses paires de petites molécules ont été simulées, prédisant un produit N-hétérocycle fluoré deux fois au niveau du carbone alpha. (À droite) Le cadre de réaction réussi utilisant la pyridine et des exemples des types de composés produits obtenus. Crédit :Synthèse de la nature (2022). DOI :10.1038/s44160-022-00128-y
Les simulations informatiques sont le plus souvent utilisées comme guide afin que les chimistes puissent déterminer plus efficacement les détails exacts d'une idée de réaction générale qu'ils ont en tête, tout comme une boussole aide à guider efficacement un explorateur vers une destination sur sa carte. Cependant, les chercheurs de l'ICReDD ont poussé les choses un peu plus loin et ont utilisé des simulations pour produire l'idée générale d'une réaction totalement inimaginable, en utilisant efficacement des calculs pour créer la carte elle-même. En utilisant le principe de conception suggéré par les résultats informatiques, l'équipe a atteint le fil conducteur du laboratoire, développant avec succès une suite de 48 réactions qui produisent des composés potentiellement utiles pour le développement de nouveaux médicaments.
La présence et la position du fluor dans une molécule affectent souvent l'activité pharmacologique d'une molécule. Des chercheurs de l'ICReDD ont utilisé des calculs de chimie quantique pour découvrir une réaction qui ajoute sélectivement deux atomes de fluor à une position difficile d'accès sur un N-hétérocycle - des molécules avec une structure cyclique de carbone où au moins un carbone de l'anneau est remplacé par de l'azote . La capacité d'attacher des atomes de fluor au "carbone alpha" auparavant difficile d'accès - le carbone immédiatement à côté de l'azote dans la structure cyclique - pourrait conduire au développement d'une multitude de nouveaux médicaments.
Avant de réaliser des expériences en laboratoire, les chercheurs ont jeté un large filet, testant par ordinateur la viabilité de nombreuses réactions à 3 composants à l'aide de la méthode de réaction induite par la force artificielle (AFIR). Ils ont simulé la réaction d'une molécule de difluorocarbène, qui agit à la source des atomes de fluor, avec différentes paires de petites molécules présentant une double ou triple liaison. Ces simulations ont montré qu'un certain nombre de réactions de formation d'anneaux devraient être viables.
Les chercheurs ont essayé l'une des réactions prometteuses suggérées par les premiers résultats de calcul, mais n'ont pas réussi. A more narrowly focused, optimized computation of the transition state energy of the reaction in question showed that the difluorocarbene molecule more easily reacted with itself than with the desired starting molecules, signaling that an undesired side reaction was likely occurring. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in Nature Synthesis . Hitting rewind to predict multi-step chemical reactions