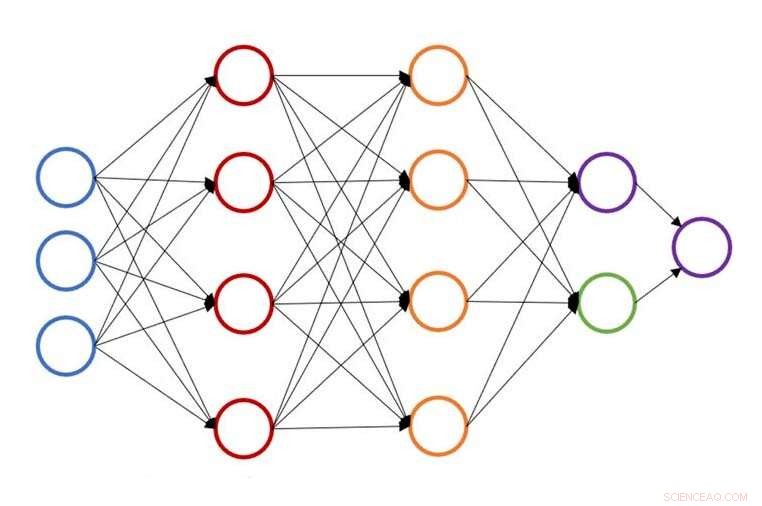
Illustration d'une structure de réseau de neurones unique pour la prédiction de l'énergie atomique. Crédit :John Kitchin, L'université de Carnegie Mellon
Apprentissage automatique, une méthode d'analyse de données utilisée pour automatiser la construction de modèles analytiques, a remodelé la façon dont les scientifiques et les ingénieurs mènent la recherche. Une branche de l'intelligence artificielle (IA) et de l'informatique, la méthode repose sur un grand nombre d'algorithmes et de vastes ensembles de données pour identifier des modèles et prendre des décisions de recherche importantes.
Des applications des techniques d'apprentissage automatique émergent dans le domaine de la catalyse de surface, permettant des simulations plus poussées de nanoparticules, études de ségrégation, optimisation des structures, apprentissage à la volée des champs de force, et criblage à haut débit. Cependant, travailler sur de grandes quantités de données peut souvent être une tâche longue et coûteuse en calcul.
Optimisation de la géométrie, souvent l'étape limitante dans les simulations moléculaires, est un élément clé des matériaux informatiques et de la science des surfaces. Il permet aux chercheurs de trouver des structures atomiques à l'état fondamental et des voies de réaction, propriétés utilisées pour estimer les propriétés cinétiques et thermodynamiques des structures moléculaires et cristallines. Bien que vital, le processus peut être relativement lent, nécessitant un grand nombre de calculs.
À l'Université Carnegie Mellon, John Kitchin s'efforce d'accélérer ce processus en fournissant une méthode d'apprentissage actif basée sur un réseau de neurones qui accélère l'optimisation géométrique pour plusieurs configurations simultanément. Le nouveau modèle réduit le nombre de calculs de la théorie fonctionnelle de la densité (DFT) ou de la théorie des milieux effectifs (EMT) de 50 à 90 %, permettant aux chercheurs de faire le même travail en moins de temps ou plus de travail dans le même temps.
"Normalement, quand on fait de l'optimisation géométrique, on part de zéro, " a déclaré Kitchin. " Les calculs bénéficient rarement de tout ce que nous savions dans le passé. "
"En ajoutant un modèle de substitution au processus, nous lui avons permis de s'appuyer sur des calculs antérieurs, plutôt que de recommencer à zéro à chaque fois."
L'étude illustre l'accélération sur plusieurs études de cas, y compris les surfaces avec adsorbats, surfaces métalliques nues, et bande élastique coudée pour deux réactions. Dans chaque cas, le package Python d'optimisation de l'environnement de simulation atomique (ASE) permettait moins de calculs DFT que la méthode standard.
Le package Python ASE-optimizer a été mis à la disposition de collègues ingénieurs et scientifiques pour faciliter l'utilisation de l'apprentissage actif des ensembles de réseaux neuronaux pour l'optimisation de la géométrie.