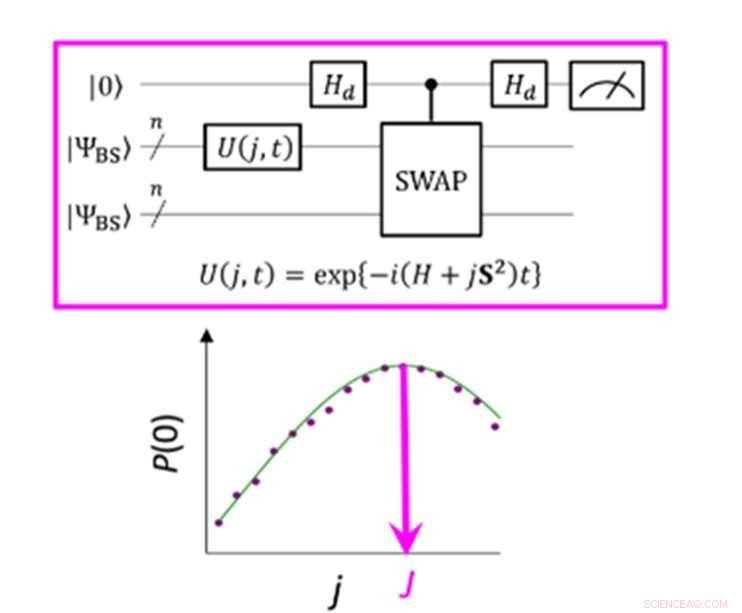
Un circuit quantique qui permet la probabilité maximale de P(0) dans la mesure du paramètre J. Crédit :K. Sugisaki, K. Sato et T. Takui
Des chercheurs de l'Université de la ville d'Osaka utilisent des états de superposition quantique et l'inférence bayésienne pour créer un algorithme quantique, facilement exécutable sur les ordinateurs quantiques, qui calcule avec précision et directement les différences d'énergie entre les états électroniques de base et de spin excité des systèmes moléculaires en temps polynomial.
Comprendre comment fonctionne le monde naturel nous permet de l'imiter pour le bien de l'humanité. Pensez à combien nous comptons sur les batteries. Au cœur se trouve la compréhension des structures moléculaires et du comportement des électrons en leur sein. Le calcul des différences d'énergie entre les états électroniques de base et de spin excité d'une molécule nous aide à comprendre comment mieux utiliser cette molécule dans une variété de produits chimiques, applications biomédicales et industrielles. Nous avons fait beaucoup de progrès dans les molécules avec des systèmes à coque fermée, dans lequel les électrons sont appariés et stables. Systèmes à coque ouverte, d'autre part, sont moins stables et leur comportement électronique sous-jacent est complexe, et donc plus difficile à comprendre. Ils ont des électrons non appariés dans leur état fondamental, qui font varier leur énergie en raison de la nature intrinsèque des spins électroniques, et rend les mesures difficiles, d'autant plus que les molécules augmentent en taille et en complexité. Bien que ces molécules soient abondantes dans la nature, il y a un manque d'algorithmes qui peuvent gérer cette complexité. L'un des obstacles a été de faire face à ce qu'on appelle l'explosion exponentielle du temps de calcul. En utilisant un ordinateur conventionnel pour calculer comment les spins non appariés influencent l'énergie d'une molécule à coque ouverte, il faudrait des centaines de millions d'années, temps que les humains n'ont pas.
Les ordinateurs quantiques sont en cours de développement pour aider à réduire ce temps à ce qu'on appelle le "temps polynomial". Cependant, le processus que les scientifiques ont utilisé pour calculer les différences d'énergie des molécules à coque ouverte a été essentiellement le même pour les ordinateurs conventionnels et quantiques. Cela entrave l'utilisation pratique de l'informatique quantique dans les applications chimiques et industrielles.
"Les approches qui invoquent de véritables algorithmes quantiques nous aident à traiter les systèmes à coque ouverte beaucoup plus efficacement qu'en utilisant des ordinateurs classiques, " déclarent Kenji Sugisaki et Takeji Takui de l'Université d'Osaka. Avec leurs collègues, ils ont développé un algorithme quantique exécutable sur des ordinateurs quantiques, qui peut, pour la première fois, calculer avec précision les différences d'énergie entre le sol électronique et les états de spin excité des systèmes moléculaires à coque ouverte. Leurs conclusions ont été publiées dans la revue Sciences chimiques le 24 décembre 2020.
La différence d'énergie entre les états de spin moléculaire est caractérisée par la valeur du paramètre d'interaction d'échange J. Les algorithmes quantiques conventionnels ont pu calculer avec précision les énergies des molécules à coque fermée "mais ils n'ont pas été capables de gérer des systèmes avec une forte multi-configuration personnage, " précise le groupe. Jusqu'à présent, les scientifiques ont supposé que pour obtenir le paramètre J, il fallait d'abord calculer l'énergie totale de chaque état de spin. Dans les molécules à coque ouverte, cela est difficile car l'énergie totale de chaque état de spin varie considérablement à mesure que la molécule change d'activité et de taille. Cependant, "la différence d'énergie elle-même ne dépend pas beaucoup de la taille du système, " note l'équipe de recherche. Cela les a amenés à créer un algorithme avec des calculs axés sur la différence de spin, pas les états de spin individuels. La création d'un tel algorithme nécessitait qu'ils abandonnent les hypothèses développées à partir d'années d'utilisation d'ordinateurs conventionnels et se concentrent sur les caractéristiques uniques de l'informatique quantique, à savoir les « états de superposition quantique ».
"Superposition" permet aux algorithmes de représenter deux variables à la fois, ce qui permet ensuite aux scientifiques de se concentrer sur la relation entre ces variables sans avoir besoin de déterminer d'abord leurs états individuels. L'équipe de recherche a utilisé ce qu'on appelle une fonction d'onde à symétrie brisée comme une superposition de fonctions d'onde avec différents états de spin et l'a réécrite dans l'équation hamiltonienne pour le paramètre J. En exécutant ce nouveau circuit quantique, l'équipe a pu se concentrer sur les écarts par rapport à sa cible et en appliquant l'inférence bayésienne, une technique d'apprentissage automatique, ils ont apporté ces écarts pour déterminer le paramètre d'interaction d'échange J. "Des simulations numériques basées sur cette méthode ont été effectuées pour la dissociation covalente de l'hydrogène moléculaire (H
« Nous prévoyons d'installer notre calculateur de paramètres de couplage Bayesian eXchange avec le logiciel de fonctions d'onde à symétrie brisée (BxB) sur des ordinateurs quantiques à court terme équipés de dispositifs quantiques bruyants (pas de correction d'erreur quantique) à échelle intermédiaire (plusieurs centaines de qubits) (dispositifs NISQ ), tester l'utilité pour les calculs de chimie quantique de systèmes moléculaires de taille réelle. »