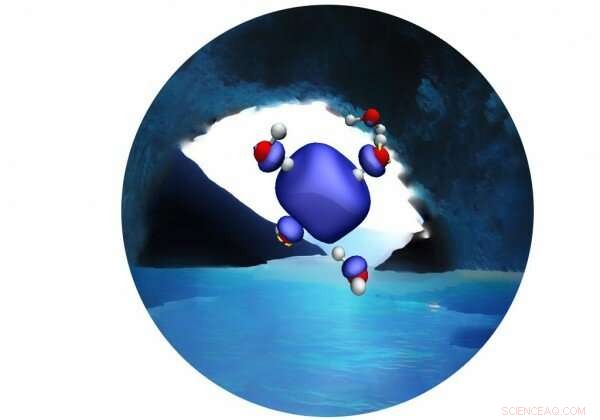
Une nouvelle simulation MD fournit des preuves concluantes en faveur d'une cavité tétraédrique persistante composée de quatre molécules d'eau. Crédit :Vladimir Rybkin
électrons supplémentaires solvatés dans l'eau liquide, appelés électrons hydratés, ont été signalés pour la première fois il y a 50 ans. Cependant, leur structure n'est pas encore bien comprise. Chercheurs MARVEL à l'Université de Zurich, L'ETH et le Centre national suisse de calcul intensif CSCS ont maintenant franchi une étape vers la résolution du mystère. Leur papier, "La dynamique de l'électron hydraté en vrac de la théorie de la fonction de nombreuses ondes corporelles, " a été publié dans Angewandte Chemie .
L'e
-
La modélisation fiable de l'électron hydraté est au moins aussi difficile que l'approche expérimentale, et les limites des approches informatiques appliquées jusqu'à présent ont conduit à une incertitude théorique considérable. Les chercheurs n'ont pas, par exemple, pu se mettre d'accord sur le fait que l'électron hydraté occupe ou non une cavité. Bien que la plupart des études théoriques suggèrent que c'est le cas, les modèles sans cavité se sont également avérés précis. Un autre point de discussion est lié aux structures de surface et de masse distinguables de l'électron hydraté.
Dans le journal, les chercheurs Vladimir Rybkin et Jan Wilhelm de l'Université de Zurich et Joost VadeVondele de l'ETH Zurich et du CSCS ont utilisé la première simulation de dynamique moléculaire de l'électron hydraté en vrac basée sur la théorie de la fonction d'onde corrélée pour fournir des preuves concluantes en faveur d'une cavité tétraédrique persistante composée de quatre molécules d'eau. Ils ont également montré qu'il n'y a pas de structures stables sans cavité dans l'électron hydraté en masse.
Les scientifiques sont arrivés à leur modèle en examinant attentivement les caractéristiques que l'approche la plus précise doit avoir. Ils voulaient qu'il soit basé sur la dynamique moléculaire pour capturer la formation et les transformations dynamiques de la cavité. Ils avaient besoin d'un niveau de structure électronique corrélé à plusieurs corps pour éviter les erreurs de délocalisation et permettre les effets de corrélation qui se sont avérés cruciaux pour prédire avec précision la solvatation de l'électron sans paramètres empiriques. Ils voulaient que la simulation soit effectuée dans le volume dans des conditions aux limites périodiques pour éviter la formation de la structure de surface et, finalement, la méthode doit fournir une description précise de l'eau liquide.
La simulation MD répond à toutes ces exigences. Il représente la première simulation dynamique d'une espèce chimique complexe en phase condensée au niveau de la fonction d'onde corrélée de la théorie. C'était la première étape critique. Le second l'exécutait en fait. De tels calculs ont été techniquement impossibles jusqu'à ce que les progrès récents, y compris ceux réalisés dans leurs propres groupes, aient permis des calculs de théorie à N corps massivement parallèles en phase condensée sur des superordinateurs de pointe tels que ceux du CSCS. Il leur a néanmoins fallu environ 1 million d'heures de nœuds sur le supercalculateur Piz Daint, le plus rapide d'Europe.
Le modèle a montré qu'une cavité se forme dans les 250 femtosecondes après l'ajout de l'excès d'électrons à l'eau liquide non perturbée. De manière critique, la simulation n'a trouvé aucune preuve de structures non cavitaires de l'électron hydraté en vrac dans des états stables ou métastables. Cela fournit des preuves théoriques beaucoup plus solides pour le modèle de cavité.