Les chercheurs de l'Université Rice et du Baylor College of Medicine ont utilisé des simulations informatiques pour étudier le processus par lequel l'hémagglutinine aide les virus à envahir et à infecter les cellules. Les chercheurs pensent que le domaine souche de la protéine se déplie et se replie dans une configuration différente lorsqu'il est déclenché, mais s'arrête pour libérer un peptide de fusion caché qui lie le virus à la cellule cible. Cliquez sur l'image pour une version plus grande. Crédit :Xingcheng Lin
Il y a un problème dans l'oscillation d'une protéine qui délivre le virus de la grippe. Les chercheurs de l'Université Rice et du Baylor College of Medicine pensent que ce mécanisme peut être une cible utile pour empêcher le virus d'infecter les cellules.
Dans un article publié dans les Actes de l'Académie nationale des sciences, l'équipe Rice-Baylor dirigée par le biophysicien José Onuchic et les biochimistes Jianpeng Ma et Qinghua Wang approfondit un complexe de glycoprotéines qu'elle a commencé à définir dans un article de 2014.
Cette protéine, hémagglutinine, se trouve à la surface des virus de la grippe et les aide à se fixer et à se déplacer à travers les membranes protectrices des cellules cibles.
L'article commence à définir le mécanisme qui permet à la protéine de se déplier et de se replier en un clin d'œil, changer sa forme pour exposer un peptide qui attache le virus à une cellule et commence l'infection. Les chercheurs pensent que les médicaments thérapeutiques peuvent utiliser ce mécanisme pour arrêter le virus.
"Cette protéine commence dans un état replié et subit une transformation globale, replier dans un état complètement différent, " dit Onuchic, co-directeur du Rice's Center for Theoretical Biological Physics (CTBP). "Mais il y a une petite partie au centre que l'évolution a conservée."
Ce seul résidu d'acide aminé conservé est l'attelage qui fait que la protéine s'arrête dans le processus de repliement. Il permet à un peptide de fusion enfoui à l'intérieur de se lier à la cellule cible et de commencer à l'infecter. Sans pause, le repliage serait trop rapide pour que la reliure ait lieu.
L'auteur principal et chercheur postdoctoral de Rice Xingcheng Lin a modélisé cette partie de la protéine, la boucle B du domaine HA2. HA2 se trouve sous un autre domaine, un capuchon connu sous le nom de HA1 qui mute pour échapper aux défenses passées. Lin a expliqué que HA1 est une cible courante pour les médicaments contre la grippe, car le domaine de coiffe exposé est plus accessible que le domaine HA2 protégé.
Le problème est que HA1 mute constamment pour résister aux médicaments, il a dit. Cela influence l'efficacité des vaccins contre la grippe chaque année. Lin et Onuchic ont déclaré que HA2 présente une meilleure cible pour les médicaments car le mécanisme est hautement conservé par l'évolution.
"Si un médicament cible HA2, le domaine ne peut pas s'échapper en faisant des mutations car les mutations elles-mêmes le rendraient non fonctionnel, ", a déclaré Lin. "Ce genre de médicament pourrait devenir un vaccin universel."
HA2 est une structure trimérique qui, lorsqu'il est déclenché par des conditions acides dans l'environnement à proximité d'une cellule cible, se transforme d'une boucle aléatoire en une bobine enroulée. Même avec la pause, il se déplie et se replie en une fraction de seconde, beaucoup trop rapide pour que les microscopes voient. Mais une simulation informatique du processus peut être ralentie.
C'est une spécialité du CTBP, qui utilise des programmes qui analysent le paysage énergétique des protéines pour prédire comment elles se replieront. Onuchic et ses collègues sont des pionniers dans la théorie selon laquelle les protéines de repliement suivent un ordre, processus « entonnoir » qui dépend de l'énergie intrinsèque de chaque atome de la chaîne, dont chacun cherche constamment son état d'énergie le plus bas. Si toutes les "perles" atomiques peuvent être identifiées, il est possible de simuler le processus de pliage complexe.
Les chercheurs de Rice utilisent souvent des modèles de protéines à gros grains, un sous-ensemble d'atomes qui représentent le tout, pour prédire comment ils se replieront. La nouvelle étude était beaucoup plus ambitieuse et visait à prédire le déroulement et le repliement complexes en utilisant non seulement chaque atome de la chaîne, mais également chaque atome de son environnement liquide, dit Onuchic.
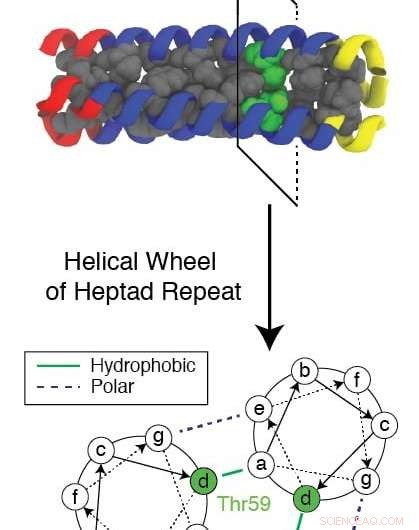
Un résidu évolutif conservé connu sous le nom de Thr59 perturbe le motif répétitif formé par une protéine trimérique lorsqu'elle se replie tout en aidant un virus de la grippe à infecter une cellule. Des chercheurs de l'Université Rice et du Baylor College of Medicine ont utilisé une simulation informatique complexe pour étudier le mécanisme et rechercher de nouvelles cibles de médicaments pour arrêter la grippe. Crédit :Xingcheng Lin
Lin a modélisé 40 microsecondes (millionièmes de seconde) de la transition du domaine HA2 qui représente l'ensemble du processus, qui prend 1,4 millisecondes (millièmes de seconde) pour terminer. Même ce processus raccourci a pris deux ans de temps informatique pour produire des résultats, il a dit.
"Le domaine simulé est d'environ 3, 000 atomes, mais quand l'environnement, y compris l'eau, est comptabilisé, la simulation totale comprend environ 100, 000 atomes, " a déclaré Onuchic. "C'est toujours une énorme simulation qui a nécessité des techniques de pointe."
Des théories antérieures basées sur des images cristallographiques des protéines avant et après ont avancé l'idée d'un domaine à ressort qui semblait s'attacher à la cellule cible après le retrait du capuchon. Onuchic a déclaré que le modèle complet de HA2 prend en charge un mécanisme différent.
"Nous avons découvert qu'il y a un tas d'énergie qui rend l'état final de HA2 beaucoup plus stable que l'état initial, " dit-il. " Mais avec le mécanisme à ressort, la majeure partie de l'énergie serait déjà gaspillée au moment où elle forme la bobine enroulée et lie les membranes cellulaires et virales. Il ne laisserait aucune énergie pour rapprocher les membranes.
"C'est pourquoi nous avons décidé de faire un calcul complet du système - tous les atomes de la protéine et toute l'eau, " a déclaré Onuchic. "C'était un effort gigantesque."
Le résidu hydrophile conservé (qui attire l'eau), connu sous le nom de Thr59, intéresse particulièrement les chercheurs non seulement pour la façon dont il perturbe le repliement et permet au virus d'attaquer, mais aussi parce qu'il a un jumeau.
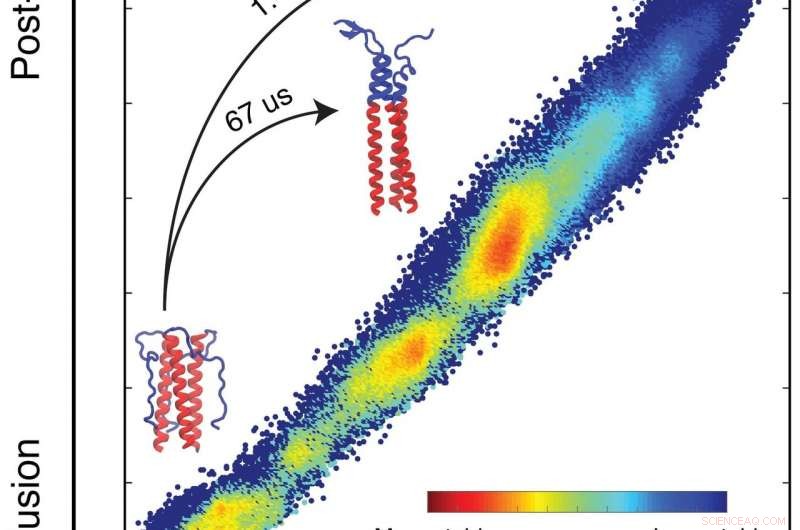
Une simulation réalisée par des biophysiciens de l'Université Rice a détaillé le profil d'énergie libre qui dicte la manière dont une protéine qui aide le virus de la grippe à infecter les cellules remplit sa mission. Les simulations prédisent comment une protéine se repliera en fonction des énergies intrinsèques de chaque atome du système. Les protéines forment des boucles et des enroulements lorsqu'elles cherchent leur plus bas, états énergétiques les plus stables (bleu). Dans le domaine étudié par les chercheurs, ils ont trouvé un problème qui ralentit le processus de pliage qui permet la liaison à la cellule cible et offre également une opportunité aux nouveaux vaccins d'attaquer la grippe. Cliquez sur l'image pour une version plus grande. Crédit :Xingcheng Lin
"Dans l'arbre évolutif complet, ces virus se divisent en deux groupes, et la différence semble être ce résidu, " a déclaré Onuchic. "Ils se sont séparés 1, Il y a 500 ans et d'une manière ou d'une autre, après cette séparation, ils sont entièrement conservés. Ils n'ont pas été en mesure de changer ce résidu quoi qu'il arrive, et nous pensons que cela rend ce résidu important."
La recherche actuelle s'est concentrée sur le groupe qui incorpore Thr59 et cause la souche H3N2 responsable de la grippe de Hong Kong, dit Lin. L'autre résidu, Met59, apparaît dans la souche H1N1 qui a causé la grippe espagnole.
"Nous avons encore un long chemin à parcourir pour comprendre l'ensemble de la protéine, " dit-il. " Tiens, nous n'avons étudié qu'un domaine d'une protéine, et il y en a plusieurs autres qui sont très importants pour sa fonction."
"Mais ce que Xingcheng a déjà fait est un tour de force informatique, " Onuchic a ajouté. " Il a montré comment ce résidu particulier brise la symétrie hélicoïdale du domaine et le rend suffisamment instable pour donner au peptide le temps de saisir les membranes. "