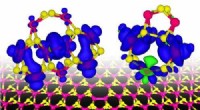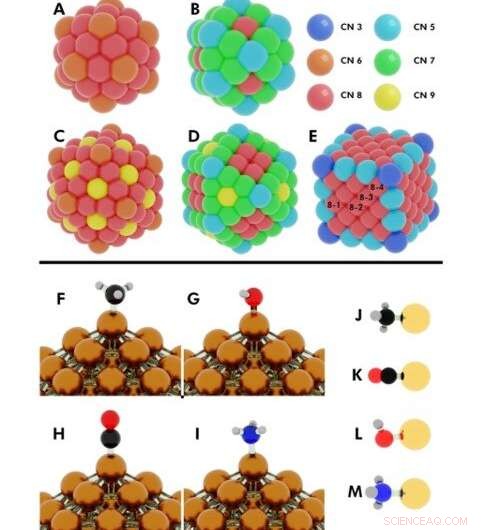
Illustration des configurations initiales de plusieurs calculs DFT (théorie fonctionnelle de la densité) effectués. En haut :nombres de coordination sur (A) icosaèdre à 55 atomes, (B) cuboctaèdre de 55 atomes, (C) icosaèdre de 147 atomes, (D) cuboctaèdre de 147 atomes, (E) Cube de 172 atomes. Nanoparticules (NPs) où plus d'un atome unique partagent le même numéro de coordination (CN), sont désignés par les numéros 8-1, 8-2, 8-3, 8-4. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aax5101
Les nanoparticules métalliques ont reçu une attention considérable en raison de leurs applications dans divers domaines de la médecine, catalyse, l'énergie et l'environnement. Cependant, les propriétés fondamentales de l'adsorption des nanoparticules sur une surface restent à comprendre. James Dean et une équipe de recherche interdisciplinaire du département de génie chimique, aux États-Unis a introduit un modèle d'adsorption universel pour tenir compte des caractéristiques structurelles, composition métallique et différents adsorbats de nanoparticules via l'apprentissage automatique (ML). Le modèle correspond à un grand nombre de données pour prédire avec précision les tendances d'adsorption sur les nanoparticules monométalliques et à base d'alliages. Le modèle était simple et fournissait des données calculées rapidement pour les métaux et les adsorbats. L'équipe de recherche a relié l'adsorption au comportement de stabilité pour faire progresser la conception de nanoparticules optimales pour les applications d'intérêt. La recherche est maintenant publiée sur Avancées scientifiques .
Les nanoparticules métalliques (NP) ont des applications importantes en catalyse, allant de la production de carburant et de produits chimiques, à l'énergie solaire et chimique. Mais leur stabilité et leur activité catalytique montrent généralement des tendances opposées, où les catalyseurs très actifs ne peuvent fonctionner que pendant quelques cycles. Une caractéristique clé de l'étendue de la fonctionnalité catalytique métallique dépend de la force d'adsorption pour une variété d'espèces sur la surface du catalyseur. Selon le principe Sabatier, développé il y a plus d'un siècle, les catalyseurs actifs doivent lier les adsorbats avec une force de liaison qui n'est ni forte ni faible. Alors que les espèces fortement adsorbées peuvent empoisonner la surface du catalyseur, les réactifs faiblement liés se désorbent facilement. Dans un scénario intermédiaire, les réactifs peuvent se rencontrer et réagir sur les surfaces catalytiques. Les chercheurs utilisent actuellement des méthodes de simulation informatique et de chimie théorique pour stimuler le comportement catalytique des catalyseurs métalliques avec une grande précision afin de guider les expériences ultérieures en laboratoire.
Les efforts de calcul se sont concentrés sur le criblage de différents catalyseurs métalliques pour découvrir l'énergie de liaison "magique" (BE) des espèces chimiques sur les surfaces des catalyseurs pour former des catalyseurs très actifs. La conception in silico de matériaux catalytiquement actifs, cependant, reste à réaliser. Les inconvénients sont principalement dus à des efforts de conception qui négligent souvent la stabilité des catalyseurs. Les catalyseurs NP possèdent également un degré élevé d'hétérogénéité de site sur leur surface pour l'adsorption et la catalyse. Les scientifiques avaient développé des modèles d'adsorption pour relier l'énergie de liaison des adsorbats aux caractéristiques de surface des NP telles que les nombres de coordination (CN) pour comprendre la réponse d'adsorption spécifique au site. Encore, pour plus de clarté, la variation de l'énergie de liaison (BE) implique également des descripteurs secondaires tels que la courbure et les propriétés électroniques des NP.
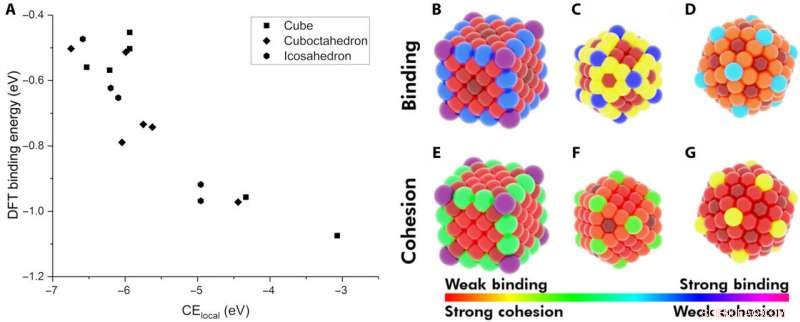
Démonstration de l'énergie de cohésion locale (CElocal) comme descripteur de l'énergie d'adsorption. (A) Le BE de CO sur différents sites de NPs Au en fonction de CElocal :cube de 172 atomes (rectangles), icosaèdre de 147 atomes (hexagones), et un cuboctaèdre de 147 atomes (losange). Carte thermique de différents sites sur les NPs par rapport à leur BE de CO (B à D) et à leur CElocal (E à G). Le schéma de couleurs suit la gamme de la liaison de CO la plus forte au CElocal le plus faible (violet) et de la liaison la plus faible au CElocal le plus fort (rouge). Crédit :Avancées scientifiques, doi:10.1126/sciadv.aax5101
Dans le travail present, Dean et al. la théorie fonctionnelle de la densité appliquée (DFT) et les techniques d'apprentissage automatique pour dériver un modèle simple basé sur la physique pour capturer avec précision l'énergie d'adsorption variable. Ils ont estimé la variable en fonction de l'environnement local du site d'adsorption à la surface du NP ainsi que du type de NP métallique. Le modèle généralisé pourrait être appliqué à n'importe quelle nanostructure métallique pour comprendre le comportement d'adsorption sur le catalyseur NP et la stabilité du catalyseur; pour cribler et concevoir des catalyseurs pour de nombreuses applications.
Les chercheurs ont d'abord émis l'hypothèse des facteurs les plus importants entre les NP monométalliques et les adsorbats. Ils ont ensuite défini l'énergie de cohésion locale (CE
The team further developed their model and performed ordinary least squares (OLS) regression to understand adsorption on monometallic NPs and slabs using three adsorbates [Methyl radical (CH

Parity plot of the model-predicted binding energy (BE) of adsorbates (OH, CO, and CH3) on various metal systems versus the DFT BE (eV). (A) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which includes the nanoparticle cohesive energy (CENP) term. (B) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which does not include the CENP term. (C) The model trained on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms) and tested against RPBE (revised Perdew-Burke-Ernzerhof model) DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu). (D) The model both trained and tested on RPBE DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu) from the slab dataset. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aax5101
Dean et al. tested the generalizability of the model and trained the simulation on a single metal or single morphology, although it accurately captured other metals or morphologies as well. The work provided strong evidence that the model captured the underlying physics of the binding interactions, allowing the team to extend the work from non-periodic NPs to periodic slab systems. Computationally inexpensive systems could parameterize the model to extend to larger systems, which was not thus far possible due to the computational costs involved.
Dean et al. then extended the model from monometallic NPs to bimetallic systems. For these experiments, they plotted the BE—trained on monometallic NPs, across several sites of bi-metallic, 55-atom icosahedron NPs (Cu
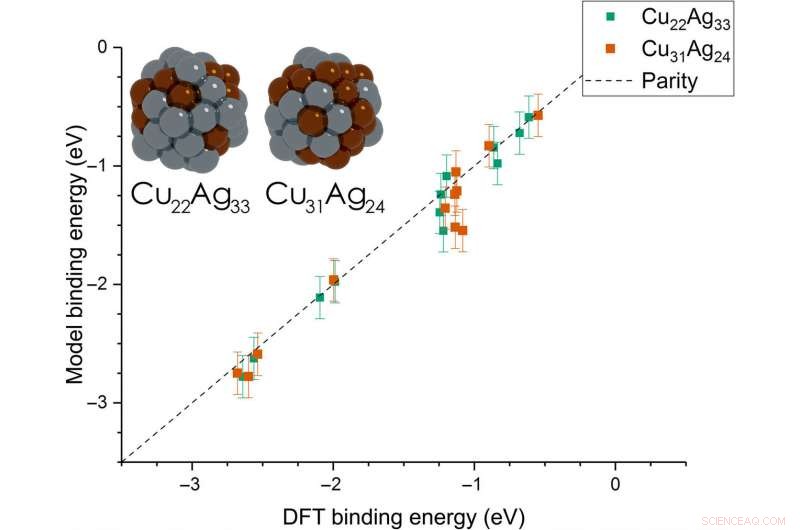
Parity plot between the presently developed model and DFT calculations on icosahedral bimetallic (Cu55−xAgx, x =24, 33) NPs. The model is trained on CH3, CO, and OH adsorbing on monometallic Ag, Cu, and Au NPs and is able to capture adsorption on bimetallic NPs. Images of the two NPs are shown as inset, with copper and silver atoms colored in brown and gray, respectivement. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aax5101
Although Dean et al. trained the ML (machine learning) algorithm to capture the adsorption trends of just one type of d9 metal, it could accurately predict the behavior of similar d9 metals (Cu—coper, Ag and Au). When they trained the model on a dataset of CH
De cette façon, James Dean and his colleagues developed a simple yet powerful physics-based model to capture trends on the strength of binding interactions between different adsorbates and metal NPs using machine learning techniques. The study was the first to develop an adsorption model that accurately connected the properties of diverse metal NPs with the stability of the adsorption site. The model introduced simple descriptors to capture the adsorption on any site, relative to monometallic and bimetallic NPs. The team generalized the model to effectively stimulate a range of binding interactions, including variations on the types of metals, their composition, sites of adsorption and adsorbates.
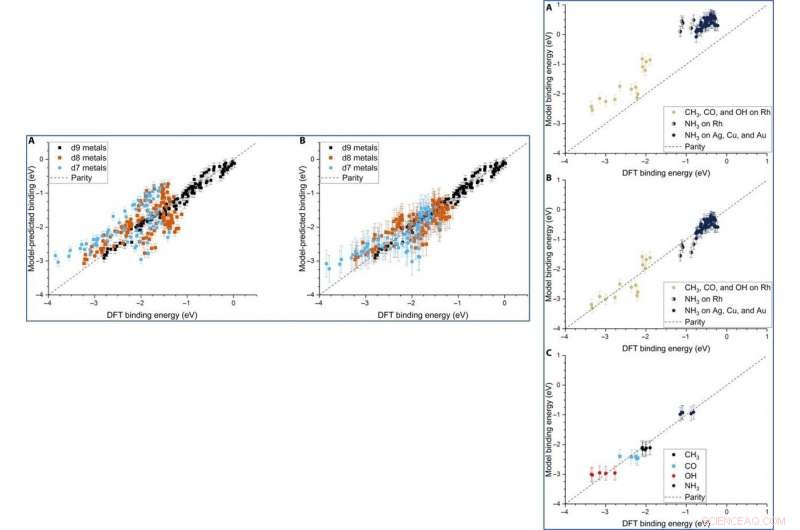
LEFT:The three-descriptor model extended to slab dataset. (A) The model trained on the slab dataset on Cu, Ag, and Au surfaces and tested against the Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au surfaces from the slab dataset. (B) The equivalent model when trained separately for each column of the d-block, still using the slab dataset. Error bars in every case are the 10-fold cross-validated RMSE of the training set. RIGHT:Extension of the model to Rh and NH3. (A) The model parameterized on our Ag, Cu, and Au NPs adsorbing CH3, CO, and OH and tested against Rh and NH3. (B) The equivalent model with empirical (constant) corrections for Rh and NH3. In the case of NH3 bound to Rh, both corrections are simultaneously applied and indicated by two-colored dots. (C) The model trained on CH3, CO, OH, and NH3 adsorbing on icosahedral/cuboctahedral Rh55. Crédit :Avancées scientifiques, doi:10.1126/sciadv.aax5101
Although the team did not test the applicability of the model for ternary systems, the physical properties may remain relevant to accurately model multimetallic systems as well. The adsorption model can accurately describe the binding strength of a variety of molecules on any site of NPs, including alloys. The scientists expect the model to be highly applicable as a screening tool for the high throughput search of potential catalysts.
© 2019 Réseau Science X