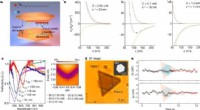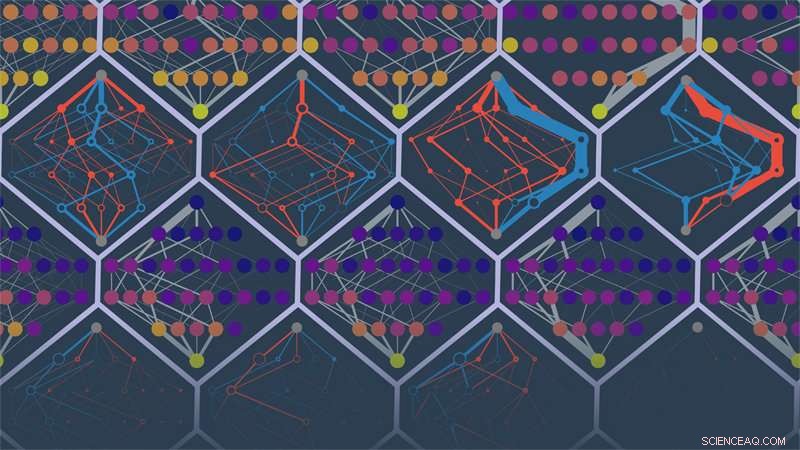
Des chercheurs du laboratoire Michael Harms de l'Université de l'Oregon ont prédit les voies évolutives de différentes protéines simulées. Les points dans l'image sont des séquences de protéines. Les arêtes indiquent les probabilités de trajectoires. Les couleurs indiquent une forme physique faible à élevée (violet à jaune) ou des itinéraires faussement inaccessibles ou faussement accessibles (rouge et bleu). Crédit :Michael Harms et Zach Sailer
Des scientifiques de l'Université de l'Oregon ont émis l'hypothèse qu'ils pouvaient manipuler une protéine une mutation à la fois et prédire son évolution. Ils ont cherché à le prouver. Et échoué.
Ils pensent, cependant, qu'ils ont trouvé une vérité fondamentale sous-jacente à l'imprévisibilité d'un système biologique. Les limitations physiques de base font de l'incertitude la norme, ils ont rapporté dans un article publié en ligne le 23 octobre dans le Actes de l'Académie nationale des sciences .
"Bien que nous ayons obtenu un résultat négatif surprenant, nous avons pu dire pourquoi, " a déclaré Michael J. Harms, professeur au Département de chimie et de biochimie de l'UO et scientifique à l'Institut de biologie moléculaire. « C'est positif. Notre simple étude confirme ce que de nombreuses personnes sur le terrain ont observé à plusieurs reprises :l'imprévisibilité. Il semble qu'elle soit universelle ».
La recherche était une affaire numérique, réalisé avec des simulations informatiques conçues par le doctorant de l'UO Zachary R. Sailer. Lui et Harms ont créé une simple protéine de réseau, en utilisant une approche préalablement créée dans le laboratoire Harms, avec une séquence aléatoire de 12 acides aminés. Ils ont ensuite effectué des simulations évolutives pour optimiser la stabilité, une propriété physique de la protéine.
L'objectif était d'utiliser les effets des 228 mutations connues pour être associées à la protéine de départ pour prédire ces trajectoires simulées :quelle mutation se produirait, lorsque, heures supplémentaires. La capacité de se projeter s'est rapidement estompée après les deux premières mutations. Après ça, les trajectoires anticipées se sont égarées au milieu d'un nombre croissant de probabilités de réacheminement.
« La qualité de vos informations se détériore avec le temps, " dit Sailer. " Au fur et à mesure que les mutations s'accumulent, les effets des mutations que vous avez mesurées commencent à changer de sorte que vous ne pouvez pas prédire où vous allez."
Dans leur papier, Sailer et Harms suggèrent que la physique, en particulier la thermodynamique, est en jeu. Chaque mutation modifie la protéine dans un petit mais manière non linéaire. Cela signifie que l'effet de chaque mutation dépend de toutes les mutations qui se sont produites auparavant.
"Je pense que ce que nous avons montré, fondamentalement, est-ce que même si vous en savez beaucoup sur un système, à propos d'une protéine, vous ne pouvez pas prédire comment il évolue à cause de la physique du système, " a dit Harms. " Il y a des règles physiques qui limitent l'évolution et sa prévisibilité. "
Comment les protéines évoluent est une question fondamentale en biologie évolutive, à la fois d'un point de vue philosophique, pour en savoir plus sur la machinerie des systèmes biologiques, et pour des indices qui pourraient conduire à des médicaments améliorés ou meilleurs.
"Pratiquement, " Harms dit, "notre recherche peut nous aider à apprendre comment prévenir l'évolution de la résistance aux antibiotiques chez les bactéries." Presque toutes les infections d'origine bactérienne développent une résistance aux antibiotiques, créant un problème de santé publique de premier plan dans le monde.
"Plutôt que d'étudier les effets de toutes les mutations individuelles, " Sailer a ajouté, "peut-être devrions-nous étudier des combinaisons aléatoires de nombreuses mutations. Une telle approche pourrait nous aider à prédire l'évolution de la résistance."
Des travaux sont en cours dans le laboratoire Harms pour tester cette possibilité sur de vraies protéines. « Nous construisons des outils informatiques qui nous permettent d'analyser des ensembles de données sur la résistance aux antibiotiques, et nous obtenons des indices qu'une approche combinatoire fonctionne, " dit Harms. " C'est plus compliqué que d'étudier des mutations individuelles, mais notre travail montre qu'il est peu probable que l'approche individuelle fonctionne."